NEJM
|
Sunil R. Lakhani, M.D.
A man cannot become a competent surgeon without the full knowledge of human anatomy and physiology, and the physician without physiology and chemistry flounders along in an aimless fashion, never able to gain any accurate conception of disease, practicing a sort of popgun pharmacy, hitting now the malady and again the patient, he himself not knowing which.
� Sir William Osler (1849�1919)
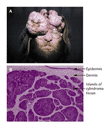
In patients with familial cylindromatosis, or "turban tumors," numerous benign skin adnexal tumors develop, principally on the head and neck (Figure 1). This rare disorder is caused by a mutation of the CYLD gene and has a variable penetrance. Thanks to three recent studies2,3,4 and, in particular, to the work of Brummelkamp and colleagues,2 we now have a more accurate conception of the disease process and thus a hope of developing a mechanism-driven therapy.
|
In the normal course of cellular homeostasis, proteins are inactivated by the attachment of ubiquitin moieties (a process called ubiquitination). These moieties act as tags by which the protein-transport machinery ferries the proteins to the proteosome for degradation. Antagonizing this process are enzymes that remove ubiquitin from proteins; CYLD is an example of a de-ubiquitinating enzyme.
Since there was already some evidence implicating de-ubiquitinating enzymes
in tumor formation, Brummelkamp et al.2 set out to
identify those that might modulate the tumor necrosis factor ![]() (TNF-
(TNF-![]() ) pathway � a key
pathway of tumor formation. They identified sequences specific to
various de-ubiquitinating enzymes and then designed a set of
sequence-specific molecules (silencing RNAs) to block their
expression. Suppression caused an increase in the level of nuclear
factor
) pathway � a key
pathway of tumor formation. They identified sequences specific to
various de-ubiquitinating enzymes and then designed a set of
sequence-specific molecules (silencing RNAs) to block their
expression. Suppression caused an increase in the level of nuclear
factor ![]() B (NF-
B (NF-![]() B)
� a key transcription factor that sits at the end of the TNF-
B)
� a key transcription factor that sits at the end of the TNF-![]() pathway and inhibits apoptosis. Thus, the authors found that CYLD
puts the brakes on NF-
pathway and inhibits apoptosis. Thus, the authors found that CYLD
puts the brakes on NF-![]() B,
indicating that the loss of CYLD may promote tumor formation by
increasing the levels of NF-
B,
indicating that the loss of CYLD may promote tumor formation by
increasing the levels of NF-![]() B and
thereby preventing cell death.
B and
thereby preventing cell death.
How does this discovery translate into potential therapy? A closer
look at the TNF-![]() pathway suggests
that CYLD and aspirin have a similar effect on NF-
pathway suggests
that CYLD and aspirin have a similar effect on NF-![]() B
levels, although they act on different parts of the pathway. When TNF-
B
levels, although they act on different parts of the pathway. When TNF-![]() binds its receptor, a molecule called TNF-receptor�associated
factor 2 (TRAF-2) binds to the cytoplasmic end of the receptor, where
it is ubiquitinated (Figure 2). Ubiquitination of
TRAF-2 leads to the activation of the inhibitor of
binds its receptor, a molecule called TNF-receptor�associated
factor 2 (TRAF-2) binds to the cytoplasmic end of the receptor, where
it is ubiquitinated (Figure 2). Ubiquitination of
TRAF-2 leads to the activation of the inhibitor of ![]() B
kinase complex, which in turn leads to the activation of NF-
B
kinase complex, which in turn leads to the activation of NF-![]() B,
and, thus, to cell survival. CYLD acts like a brake near the
beginning of the pathway � it de-ubiquitinates TRAF-2 and thus
prevents the activation of the inhibitor of
B,
and, thus, to cell survival. CYLD acts like a brake near the
beginning of the pathway � it de-ubiquitinates TRAF-2 and thus
prevents the activation of the inhibitor of ![]() B
kinase complex (and hence of NF-
B
kinase complex (and hence of NF-![]() B).
Mutation of the CYLD gene is analogous to faulty brakes on a
car, but instead of a pileup of cars, a pileup of cells results.
B).
Mutation of the CYLD gene is analogous to faulty brakes on a
car, but instead of a pileup of cars, a pileup of cells results.
|

Aspirin and its derivatives represent another brake: they inhibit the release of NF-
Source Information
From the Breakthrough Breast Cancer Research Centre, Institute of Cancer Research, and the Royal Marsden Hospital � both in London.
References
- Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000;25:160-165. [CrossRef][ISI][Medline]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003;424:797-801. [CrossRef][ISI][Medline]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003;424:793-796. [CrossRef][ISI][Medline]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003;424:801-805. [CrossRef][ISI][Medline]