NEJM
|
Patricia D. Wilson, Ph.D.
Polycystic kidney diseases are a leading cause of end-stage renal failure and a common indication for dialysis or renal transplantation. Recent advances have led to insights into mechanisms underlying the cause and prognosis of these diseases and suggest new directions for treatment.
Polycystic kidney disease may arise sporadically as a developmental abnormality or may be acquired in adult life, but most forms are hereditary. Among the acquired forms, simple cysts can develop in kidneys as a consequence of aging; dialysis, drugs, and hormones can cause multicystic disease1,2; and renal cysts are often secondary manifestations of genetic proliferative syndromes.1 The inherited polycystic kidney diseases, which are due to germ-line mutations in single genes, inherited as mendelian traits, include autosomal dominant and autosomal recessive polycystic kidney disease, nephronophthisis, and medullary cystic diseases. The age at onset, the severity of symptoms, and the rates of progression to end-stage renal failure or death vary widely in this group of diseases.
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease, the most common form of polycystic kidney disease, occurs in 1 in 800 live births. It affects 500,000 persons in the United States and 4 million to 6 million worldwide and is the reason for hemodialysis in 7 to 10 percent of patients. There are two types: type I is caused by mutations in the PKD1 gene and accounts for 85 to 90 percent of cases,3 and type II is caused by mutations in the PKD2 gene and accounts for 10 to 15 percent of cases4 (Table 1). The protein products of these two genes, polycystin-1 (Figure 1A) and polycystin-2 (Figure 1B), occur on renal tubular epithelia. Polycystin-1 is a membrane receptor capable of binding and interacting with many proteins, carbohydrates, and lipids and eliciting intracellular responses through phosphorylation pathways, whereas polycystin-2 is thought to act as a calcium-permeable channel. The two types of autosomal dominant polycystic kidney disease have similar pathological and physiological features, but type II disease has a later onset of symptoms and a slower rate of progression to renal failure; thus, patients have a longer life expectancy (69.1 years) than those with type I disease (53.0 years).6 Some patients with typical features of autosomal dominant polycystic kidney disease have no mutations in PKD1 or PKD2, suggesting that there may be a rare third form of the disease,7 although the proposed gene � PKD3 � has not been identified. Patients with mutations in both the PKD1 and PKD2 genes (transheterozygotes) have a more severe clinical course than those with mutations in only one of the genes.8
|
|

Tremendous cystic enlargement of both kidneys is characteristic of autosomal dominant polycystic kidney disease. Patients often present with hypertension, hematuria, polyuria, and flank pain and are prone to recurrent urinary tract infections and renal stones. In addition to the presence of hundreds to thousands of renal cysts, up to 10 to 20 cm in diameter, clinically significant cysts are also common in the liver (especially in women), pancreas, and intestine. Patients have an increased risk of aortic aneurysms and heart-valve defects,9 and some kindreds have five times the risk in the general population of sudden death from ruptured intracerebral aneurysms.10
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease is much rarer than the autosomal dominant form, with an incidence of 1 in 20,000 live births, and often causes fetal or neonatal death owing to tremendous bilateral enlargement of the kidneys, impaired lung formation, and pulmonary hypoplasia. Renal failure and hepatic fibrosis develop in most babies who survive the perinatal period. The disease is characterized by the expansion and elongation of collecting tubules into multiple small cysts and by biliary dysgenesis. Mutations in the PKHD1 gene cause autosomal recessive polycystic kidney disease11,12 (Table 1 and Figure 1D). The identification of a mild form of the disease13 suggests that additional genes are involved.
Familial Nephronophthisis
Familial nephronophthisis is inherited as a recessive trait. Three distinct types � juvenile, adolescent, and infantile � are caused by mutations in the NPH1 (Figure 1C), NPH2, and NPH3 genes, respectively.14,15,16 In nephronophthisis, both kidneys are shrunken and the renal cysts are restricted to the medulla at its border with the cortex. The typical clinical manifestations are salt wasting, growth retardation, anemia, polyuria, and progressive renal insufficiency.
Medullary Cystic Kidney Disease
The two distinct types of medullary cystic kidney disease are caused by mutations in either the MCKD1 or MCKD2 gene.17,18 These diseases are also characterized by bilaterally shrunken kidneys, cysts restricted to the renal medulla, salt wasting, and polyuria. But unlike nephronophthisis, medullary cystic kidney diseases are inherited as autosomal dominant traits (Table 1), are clinically milder, and typically first appear in adulthood.
Cell Biology
In autosomal dominant polycystic kidney disease, the thousands of large, spherical cysts of various sizes throughout the cortex and medulla are derived from every segment of the nephron. The tubule wall, which is lined by a single layer of epithelial cells, expands and then rapidly closes off from the tubule of origin (Figure 2B).1 By contrast, in autosomal recessive polycystic kidney disease, the smaller, elongated cysts arise as ectatic expansions of collecting ducts and maintain contact with their nephron of origin (Figure 2C). In nephronophthisis and medullary cystic kidney disease, the cysts are restricted to the corticomedullary border and may be derived from collecting ducts and distal tubules. Despite these differences among polycystic kidney diseases, common features control the formation and enlargement of renal cysts.19
|
Proliferation and Apoptosis
A precisely controlled balance between cellular proliferation and
programmed cell death (apoptosis) is essential for normal growth and
differentiation of the kidney and maintenance of normal renal
structure after birth. These fundamental processes are disturbed in
polycystic kidneys. In both autosomal dominant and autosomal
recessive polycystic kidney disease, apoptosis is abnormally
persistent20 and can destroy much of the normal
renal parenchyma, thereby allowing cystic epithelia to proliferate. The
importance of apoptosis has been highlighted in knockout mice, in
which the inactivation of inhibitors of apoptosis (bcl-2 or
activating protein 2![]() [AP-2
[AP-2![]() ])
causes cystic kidney disease.21,22
Rodent models of polycystic kidney disease are listed in Supplementary
Appendix 1 (available with the full text of this article at www.nejm.org).
])
causes cystic kidney disease.21,22
Rodent models of polycystic kidney disease are listed in Supplementary
Appendix 1 (available with the full text of this article at www.nejm.org).
The proliferation of renal tubular epithelial cells ceases before birth, but cystic epithelia proliferate abnormally throughout life in patients with autosomal dominant polycystic kidney disease.23 Moreover, cultured epithelial cells from these patients have an increased intrinsic capacity for proliferation and survival. Several genetic manipulations that cause the proliferation of tubular epithelial cells in mice also lead to renal cystic disease24,25,26,27 (see Supplementary Appendix 1).
Epidermal growth factor (EGF) has an important role in the expansion of renal cysts. Epithelial cells from cysts from patients with the autosomal dominant form and from those with the autosomal recessive form are unusually susceptible to the proliferative stimulus of EGF. Moreover, cyst fluids from the former group of patients contain mitogenic concentrations of EGF, and this EGF is secreted into the lumens of cysts in amounts that can induce cellular proliferation.28 The overexpression and abnormal location of EGF receptors on the apical (luminal) surface of cyst-lining epithelia creates a sustained cycle of autocrine�paracrine stimulation of proliferation in the cysts28 (Figure 3).
|

Genetic experiments in mice have also shown the importance of the overexpression of EGF receptors in the formation of renal cysts,29 and this work has led to the development of specific inhibitors of the EGF-receptor tyrosine kinase, which have reduced the number of cysts and extended the life span of mice with polycystic kidney disease.30 This class of small-molecule inhibitors of tyrosine kinase is now under investigation in phase 1 and 2 clinical trials in adults with polycystic kidney disease to determine whether they slow the expansion of cysts and the decline in renal function.
EGF receptors are also expressed in the apical membranes of collecting-tubule epithelia in normal fetal kidneys31 (Figure 3). Whereas basal EGF receptors in normal adult epithelia are comprised of homodimers, the apical EGF receptors consist of heterodimers of EGF receptor and erb-b2.31 The importance of this EGF-receptor variant, erb-b2, is illuminated by the fact that renal cysts form in transgenic mice that overexpress erb-b232 and that erb-b2 inhibitors have a protective effect in vitro on cells from patients with autosomal dominant polycystic kidney disease. These observations suggest that erb-b2 inhibitors might have therapeutic value.
Additional growth factors, cytokines, and lipid factors, as well as adenosine triphosphate (ATP) and cyclic adenosine monophosphate (cAMP) in cyst fluids, have proliferative effects on epithelial cells in vitro.19,33,34,35 These factors may stimulate EGF-dependent growth of cysts.36
Secretion
The net reabsorption of fluid in the normal kidney is brought about by sodium ion gradients established by the sodium pump (Na+/K+�ATPase) in the basolateral tubular cell membrane and by multiple ion and fluid transporters and channels in apical and basolateral sites. In kidneys of patients with polycystic kidney disease, Na+/K+�ATPase is abnormally located in the apical (luminal) cell membranes of tubular epithelia (Figure 3) 37 and the Na+,K+,2Cl� symporter is misplaced to the basal surface of the epithelia.38
Molecular studies of the ![]() and
and ![]() subunits of the Na+/K+�ATPase complex have
shown that normal adult kidneys contain
subunits of the Na+/K+�ATPase complex have
shown that normal adult kidneys contain ![]() 1
1![]() 1
complexes, which are located in the basolateral region of the tubule,
whereas the kidneys of patients with polycystic kidney disease
contain
1
complexes, which are located in the basolateral region of the tubule,
whereas the kidneys of patients with polycystic kidney disease
contain ![]() 1
1![]() 2
complexes in the apical membrane39 (Figure
3). In the normal fetus, Na+/K+�ATPase is
also composed of
2
complexes in the apical membrane39 (Figure
3). In the normal fetus, Na+/K+�ATPase is
also composed of ![]() 1
1![]() 2
complexes and occurs on the apical membranes of renal tubules.40
It seems that in autosomal dominant polycystic kidney disease, a
failure to down-regulate the transcription of the
2
complexes and occurs on the apical membranes of renal tubules.40
It seems that in autosomal dominant polycystic kidney disease, a
failure to down-regulate the transcription of the ![]() 2
isoform after birth facilitates the erroneous placement of Na+/K+�ATPase
in the apical membrane.
2
isoform after birth facilitates the erroneous placement of Na+/K+�ATPase
in the apical membrane.
Additional transport-related features of cysts include the presence of aquaporin 1 or aquaporin 2 water channels in cyst epithelia from patients with autosomal dominant polycystic kidney disease and of aquaporin 2 alone in cysts from patients with the autosomal recessive form.41 The high levels of ATP released by apical membranes in patients with the autosomal dominant form may further exacerbate secretion.42 Intracellular cAMP levels are also important regulators of secretion in cysts and regulate the cystic fibrosis transmembrane conductance regulator chloride channels in the apical membranes of cystic epithelia from patients with autosomal dominant polycystic kidney disease.43
Cell�Matrix Interactions
Abnormalities in the basement-membrane structure, interstitial matrix composition, the levels of matrix metalloproteases and their inhibitors, and the expression of integrin receptors occur in patients with polycystic kidney disease.40 Thickened basement membranes, alterations in matrix composition, and abnormal numbers of integrin receptors are frequent in autosomal dominant as well as autosomal recessive polycystic kidney disease and juvenile nephronophthisis. These alterations cause marked functional disturbances. For example, epithelia from patients with autosomal dominant polycystic kidney disease are more adherent to matrixes made up of collagen type I or IV than are normal epithelia and have decreased migratory capacities against growth factor gradients.44,45 Such defects may impair the cell movements required for morphogenesis of the kidney.
Genetic experiments in mice have shown that inactivation of several
matrix adhesion receptors and focal adhesion complex�associated proteins
causes the formation of cysts.46,47,48,49
Similarly, overexpression of polycystin-1 or ![]() -catenin
(Wnt) causes cysts.50,51
The migratory defect of epithelial cells from patients with autosomal
dominant polycystic kidney disease can be reversed by inhibitors of
the Wnt pathway.45 These molecules may have potential
therapeutic applications.
-catenin
(Wnt) causes cysts.50,51
The migratory defect of epithelial cells from patients with autosomal
dominant polycystic kidney disease can be reversed by inhibitors of
the Wnt pathway.45 These molecules may have potential
therapeutic applications.
Polarity
The normal adult nephron is a segmented structure lined by at least 15 distinct types of highly polarized epithelia. The polarized distribution of enzymes, ion transporters, channels, pores, growth factor, and matrix receptors facilitates normal vectorial function (directional transport) and the control of cell division, differentiation, and maturation. In autosomal dominant and autosomal recessive polycystic kidney disease, alterations in the polarity of membrane proteins include aberrant location of Na+/K+�ATPase, EGF receptors, cathepsin B, matrix metalloproteinase 2, and E-cadherin in the apical-cell membrane rather than the basolateral membrane.19,52 Polarization of proteins occurs during the maturation of nephrons in utero and proceeds by means of the regulated switching of gene expression. Persistent expression of fetal forms of Na+/K+�ATPase and EGF receptors suggest the presence of a block in the maturation program.28,31,39,40
The protein encoded by PKD1, polycystin-1, has defects in polarity and trafficking in patients with autosomal dominant polycystic kidney disease. In normal renal epithelia, polycystin-1 is confined to lateral cell membranes at sites of cell�cell adhesion (adherens junctions) and cell�matrix contacts (focal adhesions), whereas in cystic epithelia, most of this protein is intracellular.53
Signal Transduction
Many intracellular signal-transduction pathways have been implicated in the etiologic process of polycystic kidney disease. The PKD1, PKD2, and NPH1 gene products can themselves activate intracellular signaling cascades that regulate cell proliferation, migration, and differentiation35,54,55 (Figure 4). Integration of these pathways is essential for the cell movements underlying morphogenesis and the formation and maintenance of renal tubules with correct luminal diameters. Polycystins initiate these pathways through interactions with several proteins at the cell�cell adherens junctions and cell�matrix focal adhesion complexes. Loss of the focal adhesion function in mutant polycystin-1 results in the failure to recruit the focal adhesion kinase in patients with autosomal dominant polycystic kidney disease (Figure 5), and mutation of nephrocystin in nephronophthisis causes the formation of cysts.15,46 Additional regulation of the function of polycystins occurs by means of G proteins,56 phosphorylation by serine kinases57,58 or tyrosine kinases,57 dephosphorylation by receptor protein tyrosine phosphatases,59 and the intracellular second messengers calcium and cAMP.60,61,62
|
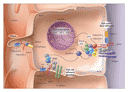
|

Current evidence suggests that complex patterns of signaling from polycystins, intracellular second messengers, and growth factors coordinate the regulation of the proliferation, differentiation, and morphogenesis of renal tubular cells through interactions with protein complexes linked to the actin cytoskeleton, intracellular signaling cascades, and the regulation of gene transcription (Figure 4). Perturbations of these regulatory mechanisms by mutations in PKD1, PKD2, or NPH1 disrupt these processes15,63,64,65 (Figure 4).
Cilia
The principal cells of the renal collecting tubule have a solitary central cilium, the function of which is obscure. Intriguingly, two mutant mouse strains with autosomal recessive polycystic kidney disease (Tg737 and cpk) have abnormal ciliary structure or function, and their encoded proteins (polaris and cystin) colocalize with polycystins in collecting-tubule cilia.66,67 However, it is not known whether these abnormalities apply to autosomal recessive polycystic kidney disease in humans, which involves a mutant PKHD1 gene. Genetic studies in a worm (Caenorhabditis elegans) and functional studies in cell culture and a Pkd-1 mutant mouse suggest a sensory role for the cilium in autosomal dominant polycystic kidney disease.68,69,70
Molecular Biology
PKD Genes and Mutations
A wide range of mutations in PKD1 or PKD2 can cause autosomal dominant polycystic kidney disease. These mutations are spread across the entire sequence of these genes and include deletions, insertions, and frame shifts and splicing, nonsense, missense, and point mutations.64 In kindreds, most mutations encode a truncated protein and are unique to a single family. PKD1, a large gene with 46 exons, encodes a 14.5-kb transcript (Table 1). The existence of additional PKD1-like homologous genes upstream of the 5' region of PKD1 complicates mutation analysis considerably. More than 100 mutations of PKD1 have been identified. By contrast, PKD2, a simpler 15-exon gene, encodes a smaller 5.6-kb transcript (Table 1). More than 75 mutations of PKD2 have been identified, again mainly of the inactivating type.71
Patients with autosomal dominant polycystic kidney disease are heterozygotes, having inherited one mutant and one normal (wild-type) allele of PKD1. A "two-hit" mechanism has been proposed to explain how cysts develop.72 This mechanism requires not only a germ-line mutation of PKD1 or PKD2 but also an additional somatic mutation in the wild-type gene to initiate the formation of cysts. Although such second hits (point mutations) do indeed occur within individual cysts, the frequencies are low (17 percent in PKD1 and up to 43 percent in PKD2),73 and polycystin protein is seen in most cyst epithelial cells of kidneys from patients with autosomal dominant polycystic kidney disease. The evidence suggests that the somatic (point) mutations must either allow transcription of the mutated wild-type allele or block production of polycystin-1 or polycystin-2.
Although complete loss of PKD1 or PKD2 clearly causes massive formation of renal cysts74 (Table 2), there is also a gene dose effect, since either too little PKD1 (complete absence or haploinsufficiency) or too much PKD1 (transgenic overexpression) causes cyst formation.50,77
|
The gene for autosomal recessive polycystic kidney disease, PKHD1, has 67 exons and encodes a large 16.2-kb transcript. Many splice variants exist, and several different mutations have been detected throughout the entire coding region. Most mutations predict the translation of a truncated protein (Table 2).
The juvenile nephronophthisis gene, NPH1, has 20 exons and encodes a small 4.5-kb transcript. Seventy to 80 percent of affected patients have homozygous deletions, whereas the remainder are heterozygotes. A variety of inactivating, nonsense, splice-site, and point mutations have been identified (Table 2).15
Polycystic Kidney Disease Proteins
Polycystin-1
Polycystin-1 is a large (>460 kD) membrane protein with a long extracellular N-terminal, 11 transmembrane domains, and a short intracellular C-terminal.75,76 Its extracellular portion contains structural motifs for binding matrix and cell-membrane proteins in the environment of renal tubular epithelia. The intracellular portion of the protein has many sites for phosphorylation and responses to regulators of signal transduction63 (Figure 1 and Figure 4). Polycystin-1 interacts and forms complexes with many other proteins in the plane of the cell membrane and on the intracellular face of the membrane that links the extracellular environment to the intracellular actin cytoskeleton (Figure 4).
Polycystin-2
The PKD2-encoded protein polycystin-2 is a 110-kD membrane protein with six transmembrane domains and intracellular N- and C-terminal domains with structural similarities to voltage-activated L-type calcium and sodium channels.4 Structural and functional analyses place polycystin-2 in the transient receptor potential family of channel proteins together with the newly identified polycystin-like calcium channels PKDL and PKD2L2.78 Although polycystin-2 can function as a nonselective cation channel that is permeable to calcium, there is uncertainty whether it functions alone or only when it forms a complex with polycystin-1 at the cell membrane or in the endoplasmic reticulum.61,79,80,81
Fibrocystin
Although little is known about the large (447-kD) protein involved in autosomal recessive polycystic kidney disease, fibrocystin (also known as polyductin), its structure suggests that it is an integral membrane receptor with extracellular protein-interaction sites and intracellular phosphorylation sites11 (Figure 1). Disease-causing mutations of PKHD1 truncate the C-terminal of fibrocystin, removing or inactivating putative signaling sites. Fibrocystin is abundant in fetal-kidney collecting ducts but absent in the kidneys of some patients with autosomal recessive polycystic kidney disease. Taken together, these properties suggest that like polycystin-1, fibrocystin may act as a membrane receptor, interacting with extracellular protein ligands and transducing intracellular signals to the nucleus.
Nephrocystin
Unlike the polycystins and fibrocystin, nephrocystin is a wholly intracellular small (83-kD) protein. It binds to focal adhesion proteins, including p130cas and tensin.15 The polycystins and nephrocystin probably interact with focal-adhesion-complex proteins, in which phosphorylation activates intracellular signaling pathways82 (Figure 4). Evidence supports the existence of a central role for the control of polycystin�nephrocystin�adhesion complexes at focal adhesion points in the cell matrix for the regulation of renal tubule geometry.
Functions of Polycystins
A wide range of analyses have led to the conclusion that polycystin-1 functions as a membrane receptor, capable of binding and interacting with many proteins, carbohydrates, and lipids83,84 and eliciting intracellular responses through phosphorylation pathways,57,85 and that polycystin-2 acts as a calcium-permeable channel.65 Polycystin-1 is found at three major renal tubular cell sites: the cell�matrix focal adhesion complex, cell�cell junctions, and the cilium, each of which links the extracellular environment of the cell membrane to the intracellular actin�tubulin cytoskeleton (Figure 4). Through multiple interactions with other proteins and modifications by phosphorylation, polycystin complexes stimulate intracellular signaling cascades that influence gene transcription.54,63,86 The evidence suggests that the polycystin complex acts as a mechanosensor, receiving signals from the extracellular matrix (by means of focal adhesions), adjacent cells (through cell junctions), and tubule lumen (through cilia), and transduces them into cellular responses that regulate proliferation, adhesion, migration, differentiation, and maturation essential for the control of the diameter of renal tubules and kidney morphogenesis.35,87
Developmental Regulation and Programming
Autosomal dominant polycystic kidney disease is a developmental disorder. Multiple renal cysts occur in utero,88 mice with homozygous targeted disruptions of the pkd1 or pkd2 gene die in utero or perinatally,49 and many fetal genes, proteins, and functions are expressed in affected patients.31,39 Polycystin-1 is developmentally regulated. There are high levels of the protein in fetal ureteric-bud and collecting-tubule cells and low levels in adult kidneys.53,89,90,91,92 In kidneys from 8-to-16-week-old fetuses, polycystin-1 is predominantly found in basal-membrane focal adhesions of migrating epithelia derived from the ureteric bud. Later in gestation and in the adult kidney, expression of polycystin-1 is down-regulated and restricted to medullary collecting tubules in lateral cell�cell adherens junctions.63 Polycystin-1 is important in the regulation of adhesion, migration, and branching elongation of the ureteric bud during renal development. Mutations in PKD1, PKD2, or NPH1 disrupt this finely coordinated process and lead to the formation of cysts.87,93
Prospects for Prognosis and Therapy
Some genetic factors that influence the rate of progression of autosomal dominant polycystic kidney disease to end-stage renal failure have been identified, including the nature of the inherited mutation. Patients with mutations in PKD2 (type II) have a slower rate of disease progression than patients with mutations in PKD1 (type I),67 mutations in the 5' portion of PKD1 lead to the early onset of a rapidly progressive disease,94,95 and the presence of both PKD1 and PKD2 mutations is predictive of severe disease.8 Associated conditions such as hypertension also increase the severity of the disease, possibly through effects of modifier genes, as suggested by the earlier onset of end-stage renal failure in patients with autosomal dominant polycystic kidney disease and angiotensin-converting�enzyme deletion polymorphisms.96,97 Another accelerator of progression is interstitial fibrosis, whether it is caused by the proliferation of fibroblasts, inflammatory cytokines, toxic and traumatic insults, or dietary additives. The recent availability of rodent models with targeted mutations in pkd1 and pkd2 or with spontaneous mutations syntenic to mutations in NPH3 (Pcy mouse)16 and PKHD1 (Pck rat)11 and well-characterized primary and conditionally immortalized renal epithelial cell cultures from normal human fetal and adult nephron segments, as well as cyst lining epithelia from patients with autosomal dominant polycystic kidney disease and autosomal recessive polycystic kidney disease,98 should speed the development of genetic, pharmacologic, or dietary interventions.
Because autosomal dominant polycystic kidney disease is slowly progressive, there is a window of opportunity to treat the disease by retarding cystic expansion. However, new methods of assessing renal decline, such as monitoring the reduction in the numbers and size of cysts, must be developed and accepted by regulatory agencies. The most promising example of potential therapy is the EGF-receptor tyrosine kinase inhibitors. Treatment of the more rapidly progressing forms of polycystic kidney diseases may present a greater challenge, since treatment will have to occur during the developmental period. In these cases, other tyrosine and serine kinase targets as well as gene-therapy approaches may prove optimal.
Source Information
From the Department of Medicine, Division of Nephrology, Mount Sinai School of Medicine, New York.
Address reprint requests to Dr. Wilson at the Department of Medicine, Division of Nephrology, Mount Sinai School of Medicine, 1425 Madison Ave., East Bldg., Rm. 11-23, New York, NY 10029, or at [email protected].
References
- Wilson PD, Falkenstein D. The pathology of human renal cystic disease. In: Dodd SM, ed. Tubulointestinal and cystic disease of the kidney. Vol. 88 of Current topics in pathology. Berlin, Germany: Springer-Verlag, 1995:1-50.
- Avner ED, Piesco NP, Sweeney WE Jr, Studnicki FM, Fetterman GH, Ellis D. Hydrocortisone-induced cystic metanephric maldevelopment in serum-free organ culture. Lab Invest 1984;50:208-218. [Abstract]
- The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994;77:881-894. [Erratum, Cell 1994;78:725, 1995;81:following 1170.] [ISI][Medline]
- Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996;272:1339-1342. [Abstract]
- Otto EA, Schermer B, Obara T, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 2003;34:413-420. [CrossRef][ISI][Medline]
- Hateboer N, van Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet 1999;353:103-107. [CrossRef][ISI][Medline]
- Ariza M, Alvarez V, Marin R, et al. A family with a milder form of adult dominant polycystic kidney disease not linked to the PKD1 (16p) or PKD2 (4q) genes. J Med Genet 1997;34:587-589. [Abstract]
- Pei Y, Paterson AD, Wang KR, et al. Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet 2001;68:355-363. [CrossRef][ISI][Medline]
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J
Med 1993;329:332-342.
[Full Text] - Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk
of rupture of intracranial aneurysms: a systematic review. Stroke
1998;29:251-256.
[Abstract/Full Text] - Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 2002;30:259-269. [CrossRef][ISI][Medline]
- Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet 2002;70:1305-1317. [CrossRef][ISI][Medline]
- Zerres K, Rudnik-Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med 1998;76:303-309. [CrossRef][ISI][Medline]
- Antignac C, Arduy CH, Beckmann JS, et al. A gene for familial juvenile nephronophthisis (recessive medullary cystic kidney disease) maps to chromosome 2p. Nat Genet 1993;3:342-345. [ISI][Medline]
- Hildebrandt F, Otto E. Molecular genetics of nephronophthisis
and medullary cystic kidney disease. J Am Soc Nephrol 2000;11:1753-1761.
[Abstract/Full Text] - Omran H, Haffner K, Burth S, et al. Human adolescent
nephronophthisis: gene locus synteny with polycystic kidney disease in pcy
mice. J Am Soc Nephrol 2001;12:107-113.
[Abstract/Full Text] - Hateboer N, Gumbs C, Teare MD, et al. Confirmation of a gene locus for medullary cystic kidney disease (MCKD2) on chromosome 16p12. Kidney Int 2001;60:1233-1239. [CrossRef][ISI][Medline]
- Fuchshuber A, Kroiss S, Karle S, et al. Refinement of the gene locus for autosomal dominant medullary cystic kidney disease type 1 (MCKD1) and construction of a physical and partial transcriptional map of the region. Genomics 2001;72:278-284. [CrossRef][ISI][Medline]
- Wilson PD. Pathogenesis of polycystic kidney disease: altered cellular function. In: Watson ML, Torres VE, eds. Polycystic kidney diseases. Oxford, England: Oxford University Press, 1996:125-63.
- Woo D. Apoptosis and loss of renal tissue in polycystic kidney
diseases. N Engl J Med 1995;333:18-25.
[Abstract/Full Text] - Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993;75:229-240. [ISI][Medline]
- Moser M, Pscherer A, Roth C, et al. Enhanced apoptotic cell
death of renal epithelial cells in mice lacking transcription factor
AP-2beta. Genes Dev 1997;11:1938-1948.
[Abstract/Full Text] - Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol 1995;5:1462-1468. [Abstract]
- Trudel M, D'Agati V, Costantini F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int 1991;39:665-671. [ISI][Medline]
- MacKay K, Striker LJ, Pinkert CA, Brinster RL, Striker GE. Glomerulosclerosis and renal cysts in mice transgenic for the early region of SV40. Kidney Int 1987;32:827-837. [ISI][Medline]
- Schaffner DL, Barrios R, Massey C, et al. Targeting of the rasT24 oncogene to the proximal convoluted tubules in transgenic mice results in hyperplasia and polycystic kidneys. Am J Pathol 1993;142:1051-1060. [Abstract]
- Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander
WS. Polycystic kidneys and chronic inflammatory lesions are the delayed
consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1).
Proc Natl Acad Sci U S A 2002;99:943-948.
[Abstract/Full Text] - Du J, Wilson PD. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol 1995;269:C487-C495. [ISI][Medline]
- Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP,
Avner ED. Epidermal growth factor receptor activity mediates renal cyst
formation in polycystic kidney disease. J Clin Invest 1998;101:935-939.
[Abstract/Full Text] - Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 2000;57:33-40. [CrossRef][ISI][Medline]
- Burrow CR, Wilson SJ, Wilson PD. Developmental regulation of Erb-b2 in human kidney organogenesis with persistent expression in autosomal dominant polycystic kidney disease. Mol Biol Cell 1997;8:Suppl:441a-441a. abstract.
- Stocklin E, Botteri F, Groner B. An activated allele of the c-erbB-2 oncogene impairs kidney and lung function and causes early death of transgenic mice. J Cell Biol 1993;122:199-208. [Abstract]
- Grantham JJ, Ye M, Davidow C, Holub BJ, Sharma M. Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995;6:1242-1249. [Abstract]
- Chatterjee S, Shi WY, Wilson P, Mazumdar A. Role of lactosylceramide and MAP kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J Lipid Res 1996;37:1334-1344. [Abstract]
- Bhunia AK, Piontek K, Boletta A, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002;109:157-168. [ISI][Medline]
- Hanaoka K, Guggino W. cAMP regulates cell proliferation and
cyst formation in autosomal polycystic kidney disease cells. J Am Soc
Nephrol 2000;11:1179-1187.
[Abstract/Full Text] - Wilson PD, Sherwood AC, Palla K, Du J, Watson R, Norman JT. Reversed polarity of Na(+)-K(+)-ATPase: mislocation to apical plasma membranes in polycystic kidney disease epithelia. Am J Physiol 1991;260:F420-F430. [ISI][Medline]
- Lebeau C, Hanaoka K, Moore-Hoon ML, Guggino WB, Beawens R, Devuyst O. BSC2 is the basolateral chloride transporter in a subset of cysts in autosomal dominant polycystic kidney disease (ADPKD). J Am Soc Nephrol 1999;10:36A-36A. abstract.
- Wilson PD, Devuyst O, Li X, et al. Apical plasma membrane
mispolarization of NaK-ATPase in polycystic kidney disease epithelia is
associated with aberrant expression of the beta2 isoform. Am J Pathol
2000;156:253-268.
[Abstract/Full Text] - Burrow CR, Devuyst O, Li X, Gatti L, Wilson PD. Expression of the beta2-subunit and apical localization of Na+-K+-ATPase in metanephric kidney. Am J Physiol 1999;277:F391-F403. [ISI][Medline]
- Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD. Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol 1996;271:F169-F183. [ISI][Medline]
- Schwiebert EM, Wallace DP, Braunstein GM, et al. Autocrine
extracellular purinergic signaling in epithelial cells derived from
polycystic kidneys. Am J Physiol Renal Physiol 2002;282:F763-F775.
[Abstract/Full Text] - Hanaoka K, Devuyst O, Schwiebert EM, Wilson PD, Guggino WB. A role for CFTR in human autosomal dominant polycystic kidney disease. Am J Physiol 1996;270:C389-C399. [Erratum, Am J Physiol 1996;271(August)(2)(Part 1):Section C following table of contents, December(6)(Part 3):Section C following table of contents.] [ISI][Medline]
- Wilson PD, Geng L, Li X, Burrow CR. The PKD1 gene product, "polycystin-1," is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest 1999;79:1311-1323. [Abstract]
- Hyink DP, Burrow CR, Bloswick B, Wilson PD. Reversal of the ADPKD epithelial cell decreased motility phenotype by lithium. J Am Soc Nephrol 2000;11:392A-392A. abstract.
- Kreidberg JA, Donovan MJ, Goldstein SL, et al. Alpha 3 beta 1
integrin has a crucial role in kidney and lung organogenesis. Development
1996;122:3537-3547.
[Abstract/Full Text] - Lo SH, Yu QC, Degenstein L, Chen LB, Fuchs E. Progressive
kidney degeneration in mice lacking tensin. J Cell Biol 1997;136:1349-1361.
[Abstract/Full Text] - Hildebrandt F, Otto E, Rensing C, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet 1997;17:149-153. [ISI][Medline]
- Lu W, Peissel B, Babakhanlou H, et al. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 1997;17:179-181. [ISI][Medline]
- Pritchard L, Sloane-Stanley JA, Sharpe JA, et al. A human PKD1
transgene generates functional polycystin-1 in mice and is associated with a
cystic phenotype. Hum Mol Genet 2000;9:2617-2627.
[Abstract/Full Text] - Saadi-Kheddouci S, Berrebi D, Romagnolo B, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 2001;20:5972-5981. [CrossRef][ISI][Medline]
- Charron AJ, Nakamura S, Bacallao R, Wandinger-Ness A.
Compromised cytoarchitecture and polarized trafficking in autosomal dominant
polycystic kidney disease cells. J Cell Biol 2000;149:111-124.
[Abstract/Full Text] - Wilson PD. Epithelial cell polarity and disease. Am J Physiol 1997;272:F434-F442. [ISI][Medline]
- Arnould T, Kim E, Tsiokas L, et al. The polycystic kidney
disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun
N-terminal kinase-dependent activation of the transcription factor AP-1. J
Biol Chem 1998;273:6013-6018.
[Abstract/Full Text] - Arnould T, Sellin L, Benzing T, et al. Cellular activation
triggered by the autosomal dominant polycystic kidney disease gene product
PKD2. Mol Cell Biol 1999;19:3423-3434.
[Abstract/Full Text] - Parnell SC, Magenheimer BS, Maser RL, et al. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun 1998;251:625-631. [CrossRef][ISI][Medline]
- Li HP, Geng L, Burrow CR, Wilson PD. Identification of phosphorylation sites in the PKD1-encoded protein C-terminal domain. Biochem Biophys Res Commun 1999;259:356-363. [CrossRef][ISI][Medline]
- Li X, Li HP, Amsler K, Hyink D, Wilson PD, Burrow CR. PRKX, a
phylogenetically and functionally distinct cAMP-dependent protein kinase,
activates renal epithelial cell migration and morphogenesis. Proc Natl Acad
Sci U S A 2002;99:9260-9265.
[Abstract/Full Text] - Sandford R, Thomas R, Greenberg A, Poon J, Boucher C, Wilson
PD. The first PKD domain of polycystin-1 interacts with a member of the
receptor protein tyrosine phosphatase protein family. J Am Soc Nephrol
2002;13:46A-46A. abstract.
[Full Text] - Geng L, Burrow CR, Li HP, Wilson PD. Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation. Biochim Biophys Acta 2000;1535:21-35. [ISI][Medline]
- Hanaoka K, Qian F, Boletta A, et al. Coassembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000;408:990-994. [CrossRef][ISI][Medline]
- Delmas P, Nomura H, Li X, et al. Constitutive activation of
G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem
2002;277:11276-11283.
[Abstract/Full Text] - Wilson PD. Polycystin: new aspects of structure, function, and
regulation. J Am Soc Nephrol 2001;12:834-845.
[Abstract/Full Text] - Harris PC. Autosomal dominant polycystic kidney disease: clues
to pathogenesis. Hum Mol Genet 1999;8:1861-1866.
[Abstract/Full Text] - Igarashi P, Somlo S. Genetics and pathogenesis of polycystic
kidney disease. J Am Soc Nephrol 2002;13:2384-2398.
[Full Text] - Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney
disease proteins, polycystin-1, polycystin-2, polaris and cystin, are
co-localized in renal cilia. J Am Soc Nephrol 2002;13:2508-2516.
[Abstract/Full Text] - Hou X, Mrug M, Yoder BK, et al. Cystin, a novel
cilia-associated protein, is disrupted in the cpk mouse model of polycystic
kidney disease. J Clin Invest 2002;109:533-540.
[Abstract/Full Text] - Barr MM, DeModena J, Braun D, Nguyen CQ, Hall DH, Sternberg PW. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr Biol 2001;11:1341-1346. [CrossRef][ISI][Medline]
- Praetorius HA, Spring KR. Bending of the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 2001;184:71-79. [CrossRef][ISI][Medline]
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003;33:129-137. [CrossRef][ISI][Medline]
- Pei Y, He N, Wang K, et al. A spectrum of mutations in the polycystic kidney disease-2 (PKD2) gene from eight Canadian kindreds. J Am Soc Nephrol 1998;9:1853-1860. [Abstract]
- Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996;87:979-987. [ISI][Medline]
- Ong AC, Harris PC, Davies DR, et al. Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int 1999;56:1324-1333. [CrossRef][ISI][Medline]
- Brook-Carter PT, Peral B, Ward CJ, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease -- a contiguous gene syndrome. Nat Genet 1994;8:328-332. [ISI][Medline]
- Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell 1995;81:289-298. [ISI][Medline]
- Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10:151-160. [ISI][Medline]
- Lu W, Fan X, Basora N, et al. Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet 1999;21:160-161. [CrossRef][ISI][Medline]
- Stayner C, Zhou J. Polycystin channels and kidney disease. Trends Pharmacol Sci 2001;22:543-546. [CrossRef][ISI][Medline]
- Koulen P, Cai Y, Geng L, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 2002;4:191-197. [CrossRef][ISI][Medline]
- Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP.
Specific association of the gene product of PKD2 with the TRPC1 channel.
Proc Natl Acad Sci U S A 1999;96:3934-3939.
[Abstract/Full Text] - Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R. The
polycystic kidney disease protein PKD2 interacts with Hax-1, a protein
associated with the actin cytoskeleton. Proc Natl Acad Sci U S A
2000;97:4017-4022.
[Abstract/Full Text] - Benzing T, Gerke P, Hopker K, Hildebrandt F, Kim E, Walz G.
Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers
phosphorylation of Pyk2. Proc Natl Acad Sci U S A 2001;98:9784-9789.
[Abstract/Full Text] - Weston BS, Bagneris C, Price RG, Stirling JL. The polycystin-1 C-type lectin domain binds carbohydrate in a calcium-dependent manner, and interacts with extracellular matrix proteins in vitro. Biochim Biophys Acta 2001;1536:161-176. [ISI][Medline]
- Malhas AN, Abuknesha RA, Price RG. Interaction of the leucine-rich
repeats of polycystin-1 with extracellular matrix proteins: possible role in
cell proliferation. J Am Soc Nephrol 2002;13:19-26.
[Abstract/Full Text] - Parnell SC, Calvet JP. Phosphorylation of the PKD1 protein, polycystin-1. J Am Soc Nephrol 1997;8:378A-378A. abstract.
- Kim E, Arnould T, Sellin LK, et al. The polycystic kidney
disease 1 gene product modulates Wnt signaling. J Biol Chem
1999;274:4947-4953.
[Abstract/Full Text] - Polgar K, Li X, Hyink DP, et al. Functional analysis of polycystin-I in renal development: microinjection of VVC lentiviral vector engineered PKD1 gene into metanephroi of mouse embryos. J Am Soc Nephrol 2002;13:112A-112A. abstract. [CrossRef]
- Sedman A, Bell P, Manco-Johnson M, et al. Autosomal dominant polycystic kidney disease in childhood: a longitudinal study. Kidney Int 1987;31:1000-1005. [ISI][Medline]
- Ward CJ, Turley H, Ong AC, et al. Polycystin, the polycystic
kidney disease 1 protein, is expressed by epithelial cells in fetal, adult,
and polycystic kidney. Proc Natl Acad Sci U S A 1996;93:1524-1528.
[Abstract/Full Text] - Geng L, Segal Y, Pavlova A, et al. Distribution and developmentally regulated expression of murine polycystin. Am J Physiol 1997;272:F451-F459. [ISI][Medline]
- Van Adelsberg J, Chamberlain S, D'Agati V. Polycystin expression is temporally and spatially regulated during renal development. Am J Physiol 1997;272:F602-F609. [ISI][Medline]
- Ong AC, Harris PC, Biddolph S, Bowker C, Ward CJ. Characterisation and expression of the PKD-1 protein, polycystin, in renal and extrarenal tissues. Kidney Int 1999;55:2091-2116. [CrossRef][Medline]
- Burrow CR. Regulatory molecules in kidney development. Pediatr Nephrol 2000;14:240-253. [ISI][Medline]
- Longa L, Scolari F, Brusco A, et al. A large TSC2 and PKD1 gene deletion is associated with renal and extrarenal signs of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997;12:1900-1907. [Abstract]
- Rossetti S, Burton S, Strmecki L, et al. The position of the
polycystic kidney disease 1 (PKD1) gene mutation correlates with the
severity of renal disease. J Am Soc Nephrol 2002;13:1230-1237.
[Abstract/Full Text] - Chapman AB, Schrier RW. Pathogenesis of hypertension in autosomal dominant polycystic kidney disease. Semin Nephrol 1991;11:653-660. [ISI][Medline]
- Baboolal K, Ravine D, Daniels J, et al. Association of the angiotensin I converting enzyme gene deletion polymorphism with early onset of ESRF in PKD1 adult polycystic kidney disease. Kidney Int 1997;52:607-613. [ISI][Medline]
- Wilson PD. In vitro methods in renal research: In: Avner ED, Harmon WE, Niaudet P, eds. Pediatric nephrology. 5th ed. Baltimore: Lippincott Williams & Wilkins, 2003:269-81.