NEJM
Medical Progress
|
The autoimmune polyendocrine syndromes are diverse, and their diversity is a characteristic that is both clinically important and instructive when their basic immunologic features are considered (Table 1).1,2,3,4 These syndromes include monogenic disorders (such as autoimmune polyendocrine syndrome type I, which has classic and characteristic disease associations5) and complex genetic disorders (such as autoimmune polyendocrine syndrome type II, in which the component diseases are more variable6 ). Some of the component disorders are common (e.g., thyroid autoimmunity and celiac disease), whereas others are rare (e.g., Addison's disease and myasthenia gravis). Some of the disorders are usually asymptomatic (e.g., celiac disease); others are symptomatic but typically diagnosed after years of illness (Addison's disease, which features severe fatigue and nausea, and pernicious anemia, which causes neuropathic symptoms); and still others are clinically dramatic at the time of diagnosis (type 1A diabetes, also known as immune-mediated diabetes and formerly called insulin-dependent diabetes). The term "polyendocrine" itself is a misnomer, in that not all patients have multiple endocrine disorders, and many have nonendocrine autoimmune diseases. Nevertheless, the recognition that patients in whom multiple autoimmune disorders are diagnosed may have a specific genetic syndrome, may be at increased risk for multiple autoimmune disorders, and may have relatives who have an increased risk should spur clinicians toward early diagnosis and treatment.7
|
A general question concerning the autoimmune polyendocrine disorders relates to the shared "antigen" that can result in the targeting of multiple tissues. In fact, it is likely that the affected organs and tissues do not share any specific molecule but rather have different molecules that are more or less likely to be targets when the immune system fails to maintain self-tolerance to a variety of molecules, in particular specific peptides within target organs (Figure 1). In addition, specific genetic polymorphisms influence which specific diseases develop; for example, a polymorphism of the insulin gene related to the thymic expression of insulin alters the risk of type 1A diabetes but not the risk of other autoimmune disorders.8
|
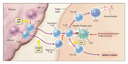
Animal Models of Pathogenesis
In the simplest hypothesis for understanding organ-specific autoimmunity,
the initial step is the loss of immunologic tolerance to a peptide
within a specific molecule found in the target organ. Clones of the
CD4 T cells that recognize the peptide then expand, and the specific
cytokines produced by the clonal CD4 T cells favor inflammation (as
when type 1 helper T [Th1]�cell clones produce cytokines such as
interferon-![]() ) or favor autoantibody-mediated
disease (as is the case predominantly with type 2 helper T [Th2]�cell
clones).9 The probability of T-cell
autoreactivity is determined both in the thymus (the site of central
tolerance) and in the periphery (the site of peripheral tolerance)
and is strongly influenced by specific HLA alleles (Figure
1).
) or favor autoantibody-mediated
disease (as is the case predominantly with type 2 helper T [Th2]�cell
clones).9 The probability of T-cell
autoreactivity is determined both in the thymus (the site of central
tolerance) and in the periphery (the site of peripheral tolerance)
and is strongly influenced by specific HLA alleles (Figure
1).
Distinct HLA alleles probably contribute to disease by determining which peptides are targeted and thus which tissue will be affected. The breakdown of self-tolerance is probably influenced by the milieu in which a peptide is presented. For example, inflammation induced by activation of the innate immune system (e.g., after infections) can favor loss of tolerance.10 Such loss of tolerance is a likely explanation for the occasional presence of autoantibodies against multiple organs as well as autoimmune disease that sometimes occurs after interferon therapy.11,12,13 The difference between a patient with a single disease and a patient with the autoimmune polyendocrine syndrome in whom one or more diseases develop over time is probably at least in part genetically determined; but it also should be recognized that disease penetrance, even among monozygotic twins, does not approach 100 percent (30 to 70 percent is usual), even in common autoimmune disorders.14,15
Genes that lower the set point for maintaining tolerance, such that T-cell expansion is more readily triggered after T-cell�receptor engagement, can cause autoimmune disorders such as the X-linked polyendocrinopathy, immune dysfunction, and diarrhea syndrome.16 Another attractive hypothesis explaining the development of autoimmunity is that genes that increase thymic expression of peripheral self antigens, thereby favoring tolerance, would decrease the risk of disease.17,18 Genes or environmental factors that increase normal T-cell regulatory function should prevent autoimmunity.19 Infections that activate the innate immune system can increase the risk of disease, whereas some infections are likely to decrease this risk, perhaps by stimulating regulatory T cells (the hygiene hypothesis).20
Studies in animal models have contributed to the understanding of polyendocrine autoimmune diseases. Three of the most important are mice that have undergone neonatal thymectomy (or thymectomy and radiation),21 the BioBreeding (BB) rat, and the nonobese diabetic (NOD) mouse. Removing the thymus from an otherwise normal, three-day-old mouse results in lymphocytic invasion of multiple organs with attendant inflammation, leading to gastritis, oophoritis, and thyroiditis.22 It is thought that in this model, which is dependent on specific timing of the thymectomy, the animal loses regulatory T lymphocytes that normally dampen the development of autoimmunity.
The NOD mouse, in which type 1A diabetes and sialitis develop spontaneously,
has more than 15 genetic loci that contribute to disease,23,24
including alleles of the major histocompatibility complex (MHC) and,
in particular, those of I-Ag7, an HLA class II molecule.25
A peptide composed of amino acids 9 through 23 of the insulin ![]() chain is recognized by the T lymphocytes of NOD mice; T cells that
are activated by the peptide then invade the pancreatic islets, and
the ultimate result is type 1A diabetes.26 This
peptide, the B:9-23 self peptide, induces insulin autoantibodies when
injected into normal BALB/c mice. When it is administered with
polyriboinosinic-polyribocytidylic acid (poly-IC, a mimic of viral
double-stranded RNA and an activator of the innate immune system),
insulitis is induced; in special strains of mice, diabetes is
induced.26,27
In the NOD mouse, once autoimmunity is activated, insulin is only one
of several islet antigens targeted by T lymphocytes; in particular,
besides insulin, another beta-cell�specific antigen (islet
glucose�related phosphatase) is a target of CD8 T cells.28,29,30
chain is recognized by the T lymphocytes of NOD mice; T cells that
are activated by the peptide then invade the pancreatic islets, and
the ultimate result is type 1A diabetes.26 This
peptide, the B:9-23 self peptide, induces insulin autoantibodies when
injected into normal BALB/c mice. When it is administered with
polyriboinosinic-polyribocytidylic acid (poly-IC, a mimic of viral
double-stranded RNA and an activator of the innate immune system),
insulitis is induced; in special strains of mice, diabetes is
induced.26,27
In the NOD mouse, once autoimmunity is activated, insulin is only one
of several islet antigens targeted by T lymphocytes; in particular,
besides insulin, another beta-cell�specific antigen (islet
glucose�related phosphatase) is a target of CD8 T cells.28,29,30
Both diabetes and thyroiditis develop spontaneously in the BB rat. Genes of the MHC are the major determinant of diabetes in the BB rat,31 but for this strain of rats an autosomal recessive mutation of a gene causes severe T-cell lymphopenia.32,33 Diabetes develops spontaneously in BB rats only if they have both lymphopenia and MHC alleles of the RT1-U type; the presence of other MHC alleles leads to thyroiditis when lymphopenia is present.34 Thus, the manifestations of autoimmune disease depend both on the type of MHC alleles and the presence or absence of lymphopenia, which is induced by a specific recessive mutation.
The most important general rule from animal models of autoimmune disease is that genes within the major histocompatibility complex, in particular immune-response genes (similar to the HLA-DQ and HLA-DR alleles in humans), are essential for disease targeting, and such targeting combined with abnormalities in immunoregulation leads to polyendocrine autoimmunity. Disease depends on cells derived from bone marrow, and major effectors are both CD4 and CD8 T lymphocytes; just as important, however, is that regulatory T lymphocytes can prevent disease.19 Because they are inbred, each animal model can be viewed as just one example of what are genetically heterogeneous disorders in humans. Models with a mutated "causative" gene identical to the mutated gene in humans (e.g., scurfy mice, which have the same genetic mutation as that causing X-linked polyendocrinopathy, immune dysfunction, and diarrhea syndrome) will probably be better guides to pathogenesis and therapy than are polygenic models, such as the NOD mouse, in which several effective therapies have not influenced progression to diabetes in humans.
Specific Clinical Syndromes
Autoimmune Polyendocrine Syndrome Type I
Autoimmune polyendocrine syndrome type I is a dramatic autoimmune syndrome with characteristic disease associations4,5 that often appear early in life, typically in infants with persistent candidal infection of the skin and mucous membranes without the systemic infection generally associated with severe immunodeficiency. The diagnosis of autoimmune polyendocrine syndrome type I is usually made later, when hypocalcemia due to hypoparathyroidism develops or Addison's disease is recognized in a young child. The syndrome is rare but has an increased prevalence in certain populations (e.g., inhabitants of Finland and Sardinia and Iranian Jews).
Mutations in an autoimmune-suppressor gene (AIRE, for autoimmune regulator), which encodes a transcription factor, cause the syndrome.35,36,37,38 Persons with any two of several specific conditions � mucocutaneous candidiasis, hypoparathyroidism, and Addison's disease � almost always have AIRE mutations. Mutations in the AIRE gene cause many autoimmune diseases, and affected patients are at risk for the development of multiple additional autoimmune diseases over time, including type 1A diabetes, hypothyroidism, pernicious anemia, alopecia, vitiligo, hepatitis, ovarian atrophy, and keratitis. Affected patients may also have diarrhea or obstipation that may be related to the destruction of gastrointestinal endocrine cells (enterochromaffin and enterochromaffin-like cells).39 Knockout of the AIRE gene in the mouse produces widespread autoimmunity, but the phenotype is relatively mild.18,40 Nevertheless, study of this mouse model has led to the hypothesis that one function of the normal AIRE gene might be to enhance the expression of "peripheral" antigens in the thymus, thereby promoting tolerance.18
After diagnosis, patients with autoimmune polyendocrine syndrome type I require close monitoring. Monitoring can help prevent illness associated with delayed diagnosis of additional autoimmune diseases (e.g., Addison's disease and hypoparathyroidism, which can develop during adulthood) as well as oral cancer, which may develop if candidiasis is not treated aggressively, and infection due to asplenism, which is present in a subgroup of patients.1
Autoimmune Polyendocrine Syndrome Type II
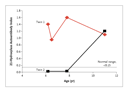
Autoimmune polyendocrine syndrome type II (also called Schmidt's syndrome with Addison's disease plus hypothyroidism) is much more common and more varied in its manifestations than autoimmune polyendocrine syndrome type I.1,3,6,41 Symptomatic hypotension, which is a classic presentation of adrenal insufficiency in autoimmune polyendocrine syndrome type II, can be associated with a decrease in the insulin dose in a patient with type 1A diabetes. Such patients may also have hyperpigmentation and vitiligo as well as a several-year history of intermittent, severe hypoglycemia and intermittent, severe fatigue. Thus, the onset is insidious until the presenting hypotensive episode (Figure 2). We are aware of one child who missed an entire year of school at the age of 10 years because of fatigue but did not receive the diagnosis of Addison's disease until the age of 17.
|
There is controversy between "splitters" and "lumpers" concerning syndrome classification.2,3,7,42 Splitters consider each of the combinations of disorders a separate syndrome: according to this approach, autoimmune polyendocrine syndrome type II refers to Addison's disease plus thyroid autoimmunity or type 1A diabetes; autoimmune polyendocrine syndrome type III refers to thyroid autoimmunity plus another autoimmunity (but not Addison's disease or type 1A diabetes); and autoimmune polyendocrine syndrome type IV refers to two or more other organ-specific autoimmune diseases. Lumpers, with whom we tend to agree, consider all the above combinations autoimmune polyendocrine syndrome type II (leaving only autoimmune polyendocrine syndrome type I and autoimmune polyendocrine syndrome type II). The disease is genetically complex, with parents, siblings, and offspring typically having multiple yet different autoimmune diseases.43,44
When a rare disorder, such as Addison's disease, occurs spontaneously, there is a high probability that other diseases are present or will develop. With a common autoimmune disorder such as hypothyroidism as the only disease, the development of additional autoimmune endocrine disorders is much less common. Diseases of intermediate prevalence, such as type 1A diabetes and celiac disease, are frequently associated with other autoimmune diseases. For example, approximately 1.5 percent of patients with type 1A diabetes have adrenal 21-hydroxylase autoantibodies, and are at high risk for Addison's disease, which develops in one third of these patients.41,43,45 It is not clear what distinguishes a patient with a single disorder, such as isolated Addison's disease, from a patient with multiple additional autoimmune disorders. One factor may simply be time, since in many patients with Addison's disease additional disorders develop with increasing age.
X-Linked Polyendocrinopathy, Immune Dysfunction, and Diarrhea
The syndrome of X-linked polyendocrinopathy, immune dysfunction, and diarrhea (known as XPID) is an extremely rare disorder characterized by fulminant, widespread autoimmunity and type 1A diabetes, which usually develops in neonates; it is often fatal.46,47 The disorder is also known as XLAAD (X-linked autoimmunity and allergic dysregulation) and IPEX (immune dysfunction, polyendocrinopathy, and enteropathy, X-linked).48,49 Recognition of the disease is important, since there is some evidence that bone marrow transplantation, with the development of mixed chimerism in the recipient, may reverse it. Studies in one patient who had a mutation in a gene called Scurfin, or FOXP3, as well as in the relevant animal model (scurfy mice), suggest that the mutated gene has an important role in setting the threshold for a response by T lymphocytes to T-cell�receptor engagement and that FOXP3 controls the development of regulatory CD4+CD25+ T cells that are essential for the maintenance of tolerance to self tissue.50
Other Syndromes
Many syndromes include multiple endocrine disorders (Table 2). Recognition of these syndromes can aid early diagnosis and permit the initiation of specific therapy. For example, administration of a sulfhydryl-containing drug such as methimazole may be stopped when it is the cause of Hirata's disease (insulin-autoimmune syndrome with insulin-autoantibody�induced hypoglycemia),51 or localized radiotherapy may be directed to a bone lesion or autologous bone marrow transplantation undertaken when the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) is present.52,53,54
|
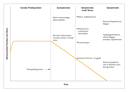
Stages in the Development of Autoimmunity with Addison's Disease
It is clear that autoimmunity precedes overt Addison's disease by years, as in many autoimmune endocrine disorders.45,55,56 Figure 3 shows stages in the hypothetical progression to Addison's disease3 and the abnormalities characteristic of each stage. Many autoimmune disorders, even non�organ-specific diseases, are likely to have similar prodromes.58,59 Hence, Addison's disease is an instructive model for end-organ failure in autoimmune polyendocrine syndromes.
|
Genetic Susceptibility
In patients with autoimmune polyendocrine syndromes who have a single disorder such as Addison's disease or type 1A diabetes, the prevalence of additional autoimmune disorders is 30 to 50 times that in the general population.60,61 The concurrence of more than one endocrinopathy presumably results from shared genetic susceptibility leading to loss of tolerance to multiple tissues.
As previously discussed, autoimmune polyendocrine syndrome type I is determined by autosomal recessive mutations in the AIRE gene. Addison's disease develops in 80 percent of patients with autoimmune polyendocrine syndrome type I, and type 1A diabetes develops in 18 percent.5,62 In contrast to autoimmune polyendocrine syndrome type I, Addison's disease � either as part of autoimmune polyendocrine syndrome type II or as an isolated condition � is a complex genetic disorder with a specific HLA-DR and HLA-DQ genotype that confers high risk. The highest risk of the development of both Addison's disease and type 1A diabetes is associated with a heterozygous HLA-DR4�DQ8/HLA-DR3�DQ2 genotype.43,44 In Addison's disease, there is an additional, strong association with a mutated allele (5.1) of a nonclassic HLA molecule, MHC class I chain-related A (MIC-A).63,64 It is likely that there are other genetic loci contributing to Addison's disease (e.g., cytotoxic T lymphocyte antigen 4),65 but identifying the disease-predisposing genes to confirm the loci has proved difficult, as it has for type 1A diabetes.66 The recent discovery of a vitiligo locus in a large family with thyroiditis67 and the ongoing analysis of a family in which 21 members have type 1A diabetes and celiac disease68 suggest that a subset of common autoimmune disorders are genetically heterogeneous with respect to key disease loci.
Triggering of Autoimmunity
A variety of factors can trigger autoimmune disease, and even a single disorder can have a variety of triggering factors. For example, administration of interferon alfa has been associated with the development of 21-hydroxylase autoantibodies, islet autoantibodies, and thyroid autoantibodies.12,13 Type 2 interferons are likely to also be important for spontaneous disease and may underlie the ability of viral infections or poly-IC, which mimics the effects of viral RNA, to induce disease in animal models.26 Triggering factors such as pregnancy may have special effects in patients with an autoimmune disease. For instance, postpartum thyroiditis develops in almost one third of patients with type 1A diabetes, and therefore patients with type 1A diabetes who are planning pregnancy should be tested for thyroid autoantibodies and monitored closely for thyroid abnormalities.69 In one third of patients with multiple sclerosis who were treated with an experimental T-cell�depleting monoclonal antibody, Graves' disease developed as a striking and unexpected complication.70
Currently, common triggering factors are known for only a very few autoimmune disorders. The most dramatic of the known triggers are the gliadin peptides, which are associated with celiac disease. Prospective studies indicate that before transglutaminase autoantibodies appear, patients may have been consuming gliadin for several years, starting in infancy, suggesting that there may be additional factors that trigger the initial activation of disease.71 An attractive hypothesis is that transglutaminase becomes a target as it deamidates glutamines of the gliadin peptides that induce disease. Gliadin peptides may also become covalently bound to transglutaminase.72
Assays for Autoimmune Markers
At present, assays for autoimmune T cells are in their infancy and not standardized,73 although important progress is being made with tetramer and enzyme-linked immunospot (Elispot) assays.74 Thus, for the earliest evidence of active autoimmunity we currently rely on assays to detect autoantibodies.45,75,76 When the autoantibody is directly pathogenic (e.g., in disorders in which transplacental autoantibodies cause disease, such as Graves' disease and myasthenia gravis), this approach is logical. For T-cell�mediated disorders, which presumably constitute most of the autoimmune diseases in which glands are destroyed, autoantibody assays are primarily disease markers. Multiple workshops held by the Immunology of Diabetes Society suggest that to achieve specificities greater than 99 percent and high sensitivity, fluid-phase radioimmunoassays are required.77 Such assays can use proteins that are transcribed and translated in vitro and simultaneously radioactively labeled; this approach is generally adaptable for assays for antibodies against most protein antigens.75,78
For the prediction of autoimmune diabetes in low-risk populations (e.g., general populations with a risk of diabetes of 1 in 300), even established radioimmunoassays indicate that people expressing a single autoantibody have a significantly lower risk than those with multiple islet autoantibodies79,80,81 (Figure 4). Autoantibodies to the enzyme 21-hydroxylase are present in more than 90 percent of patients with Addison's disease, and their detection almost always precedes the onset of disease.43,45,82 A diagnosis of adrenal insufficiency in the absence of 21-hydroxylase autoantibodies requires a search for alternative causes of Addison's disease, such as tuberculosis or adrenoleukodystrophy. Figure 2 illustrates a case in twins: high levels of 21-hydroxylase autoantibodies were present in one twin beginning at the age of six years but were not found in his identical twin until five years later as indicated by the index in the 21-hydroxylase autoantibody assay of 0.15 or greater. The autoantibody assay index is calculated as (counts per minute [cpm] with the patient's serum � cpm with normal serum) � (cpm with standard positive serum � cpm with normal serum). The first twin now has overt Addison's disease, and the second is being monitored with annual adrenal-function testing. The twins were first evaluated for 21-hydroxylase autoantibodies because they have a sibling with type 1A diabetes.
|
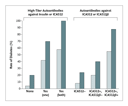
Twelve percent of patients with type 1A diabetes have transglutaminase autoantibodies.83 One third of patients with type 1A diabetes who have the HLA genotype HLA-DR3�DQ2/HLA-DR3�DQ2 have transglutaminase autoantibodies.83 Half of these patients have high levels of the autoantibodies and intestinal villous destruction, visible on examination of biopsy specimens. The levels of transglutaminase autoantibodies can fluctuate, and we recommend confirming the presence of high titers close to the time of planned biopsy. Approximately one third of patients with type 1A diabetes have thyroid autoantibodies. Chronic thyroiditis is very common, and a subgroup of patients can have thyroid dysfunction without these autoantibodies. For this reason, we monitor the level of thyrotropin. The combination of autoantibodies and a thyrotropin level greater than 5 mU per liter indicates a high risk of future development of hypothyroidism.
Progressive Metabolic Abnormalities
As illustrated in Figure 2, patients in whom Addison's disease develops have a prodrome during which 21-hydroxylase autoantibodies are present. Betterle and coworkers3 divide the progression to Addison's disease into stages, with an elevated plasma renin level as the first sign of adrenal damage. During this process, the adrenal reserve may be impaired, and we alert patients who have 21-hydroxylase autoantibodies about the symptoms and signs of adrenal insufficiency. In type 1A diabetes, the loss of first-phase insulin secretion in an intravenous glucose-tolerance test usually precedes the onset of overt diabetes; after diagnosis, loss of C-peptide secretion reflects continuing beta-cell destruction.84 In thyroiditis, the presence both of autoantibodies against thyroid peroxidase and of elevations in the thyrotropin level above the normal range increases the risk of development of overt hypothyroidism.85 Within 10 years of follow-up, approximately half the patients who are positive for autoantibodies against thyroid peroxidase and have type 1A diabetes have hypothyroidism.
Screening Recommendations
A high index of suspicion for additional autoimmune disorders in patients who have autoimmune polyendocrine syndrome type I or autoimmune polyendocrine syndrome type II, and their relatives, is essential. Patients should be advised of the symptoms of the disorders for which they are at high risk: hypoglycemia (especially when they are receiving insulin therapy), fatigue, and hyperpigmentation (in some cases) for Addison's disease; polyuria, polyphagia, polydipsia, and nausea and vomiting with ketoacidosis for diabetes; coordination difficulties for pernicious anemia; and anemia, osteopenia, abdominal pain, and diarrhea for celiac disease. (Most patients with celiac disease are not symptomatic.) We give patients a sheet of information about associated disorders.
The constellation of disorders of autoimmune polyendocrine syndrome type I relates to immunodeficiency and chronic candidiasis as well as the frequent development of additional autoimmune disorders. It is important for follow-up to include aggressive treatment of candidiasis if needed, antibiotic prophylaxis if asplenism develops (e.g., if asplenism is detected by screening for Howell�Jolly bodies), tests for early detection of hepatitis (e.g., by screening for elevated serum levels of liver enzymes), and assessment for additional endocrine disorders with autoantibody and hormone testing.1
We recommend that patients with autoimmune polyendocrine syndrome type II and their first-degree relatives (and patients with "isolated" type 1A diabetes and Addison's disease) be periodically monitored for the development of hypothyroidism. The presence of autoantibodies against thyroid peroxidase usually precedes overt hypothyroidism. Given the high prevalence of thyroid disease due to autoimmunity, we measure thyrotropin with a sensitive assay able to detect both hypothyroidism and hyperthyroidism. Pediatric endocrinologists measure thyrotropin in their patients annually, but for adults, testing at five-year intervals may be sufficient if the initial thyrotropin level is normal and autoantibodies against thyroid peroxidase are absent. If those autoantibodies are present, however, adults should be screened yearly.
We also recommend screening for autoantibodies against 21-hydroxylase and transglutaminase. Currently, there is not enough information to define optimal intervals for testing, but anecdotal data indicate that autoantibodies can develop at any age, and thus we rescreen patients for autoantibodies even if their initial autoantibody tests are negative. Annual measurement of the corticotropin level and the level of cortisol both before and after cosyntropin stimulation (at 8 a.m.) in those with autoantibodies against 21-hydroxylase seems prudent to detect adrenal damage before a hypotensive crisis.57
The majority of persons identified in programs screening for autoantibodies associated with celiac disease, even those who have high levels of transglutaminase autoantibodies, are "asymptomatic." Recent studies suggest that many of them may be at risk for osteoporosis and may have detectable changes in growth and nutrition, as well as anemia.86 Intestinal T-cell lymphomas develop in a subgroup of patients with symptomatic celiac disease. The level of transglutaminase autoantibodies often fluctuates in asymptomatic patients, and it is advisable to obtain a small-bowel biopsy specimen when the levels are high. ("High" levels are defined differently for different autoantibody assays. For example, with an autoantibody radioimmunoassay index, with 99 percent of normal persons at an assay index below 0.05, a positive biopsy finding is associated with much higher levels � namely, an index greater than 0.5.71) Islet autoantibody determination in first-degree relatives of patients with autoimmune polyendocrine syndrome type II is best reserved for research settings, although appropriate rapid diagnosis of and therapy for new-onset diabetes are important, especially in young children.87,88
Therapy
At present, the treatment of the polyendocrine autoimmune syndromes is dictated by the individual disorders. Knowledge of the syndromes allows early therapy of component disorders. An important clinical caveat is that in patients with suspected concomitant Addison's disease and hypothyroidism, thyroid replacement should not precede the necessary glucocorticoid replacement, since thyroid replacement may precipitate the hypotension and adrenal crisis due to the action of thyroxine in increasing hepatic corticosteroid metabolism. In addition, in a subgroup of patients with newly diagnosed Addison's disease who have moderately elevated levels of thyroid autoantibodies and thyrotropin levels below 30 �U per milliliter, thyrotropin levels may normalize after glucocorticoid replacement.
Obvious long-term goals are therapies that address the underlying autoimmunity associated with these disorders and, in particular, preventive therapies. Although prediction of type 1A diabetes is now possible, the preventive therapies studied to date have not altered disease progression,89 although newer approaches are under study.90,91 In contrast, for celiac disease, removal of gliadin from the diet is an effective treatment.92,93 Autoimmunity can be an important barrier to transplantation, as noted by Sutherland and coworkers in a report of findings in identical twins; transplantation of the tail of the pancreas from a normal twin to a diabetic twin was associated with recurrent diabetes in the recipient.94 With current immunosuppressive regimens, allogeneic pancreatic transplantation is possible,95 and the results of islet transplantation have improved dramatically.96 Nevertheless, a subgroup of islet transplants appear to succumb to autoimmune destruction.97
In the past decade, a wealth of new data have become available concerning the pathogenesis of rare as well as relatively common polyendocrine syndromes. It has become clear that an imbalance between autoimmune effector and regulatory T lymphocytes is the major determinant of disease, that HLA alleles determine specific tissue targeting, and that an important research goal with respect to the entire constellation of disorders is the restoration of tolerance. Currently, for the clinician and family, recognition of the syndromes and early detection of the component disorders can contribute to the prevention of illness and, in some cases, death.
Supported by grants from the National Institutes of Health
(DK32083, AI39213, DK55969, DK62718, AI95380, DK32493, DK59097,
DK63518, AI46374, and DK61926; a Diabetes Endocrine Research Center
grant [P30 DK57516]; an Autoimmunity Prevention Center grant
[AI50864]; and an Autoimmunity Center of Excellence grant
[AI046374]), a grant from the Immune Tolerance Network (AI15416),
grants from the National Institutes of Health Clinical Research
Centers Program (MO1 RR00069 and MO1 RR00051), and grants from the
American Diabetes Association, the Juvenile Diabetes Foundation, and
the Children's Diabetes Foundation.
Dr. Eisenbarth reports that he consults for Bayhill Therapeutics, Quest,
and Neurocrine and that he has stock options with Bayhill Therapeutics.
Source Information
From the Barbara Davis Center for Childhood Diabetes, University of Colorado Health Sciences Center, Denver.
Address reprint requests to Dr. Eisenbarth at the Barbara Davis Center for Childhood Diabetes, University of Colorado Health Sciences Center, 4200 E. Ninth Ave., Box B140, Denver, CO 80262, or at [email protected].
References
- Barker JM, Eisenbarth GS. Autoimmune polyendocrine syndromes. In: Eisenbarth GS, ed. Type 1 diabetes: molecular, cellular, and clinical immunology. Denver: Barbara Davis Center for Childhood Diabetes, 2003 (Web only). (Accessed April 20, 2004, at http://www.barbaradaviscenter.org.)
- Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison's disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine (Baltimore) 1981;60:355-362. [ISI][Medline]
- Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune
adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies,
autoantigens, and their applicability in diagnosis and disease prediction.
Endocr Rev 2002;23:327-364. [Erratum, Endocr Rev 2002;23:579.]
[Abstract/Full Text] - Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 1990;322:1829-1836. [Abstract]
- Perheentupa J. APS-I/APECED: the clinical disease and therapy. Endocrinol Metab Clin North Am 2002;31:295-320. [ISI][Medline]
- Schatz DA, Winter WE. Autoimmune polyglandular syndrome. II. Clinical syndrome and treatment. Endocrinol Metab Clin North Am 2002;31:339-352. [ISI][Medline]
- Eisenbarth GS, Gottlieb PA. The immunoendocrinopathy syndromes. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, eds. Williams textbook of endocrinology. 10th ed. Philadelphia: Saunders, 2003:1763-76.
- Pugliese A, Miceli D. The insulin gene in diabetes. Diabetes Metab Res Rev 2002;18:13-25. [CrossRef][ISI][Medline]
- Fields PE, Kim ST, Flavell RA. Cutting edge: changes in histone
acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2
differentiation. J Immunol 2002;169:647-650.
[Abstract/Full Text] - Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol 2002;14:380-383. [CrossRef][ISI][Medline]
- Bosi E, Minelli R, Bazzigaluppi E, Salvi M. Fulminant autoimmune Type 1 diabetes during interferon-alpha therapy: a case of Th1-mediated disease? Diabet Med 2001;18:329-332. [CrossRef][ISI][Medline]
- Wesche B, Jaeckel E, Trautwein C, et al. Induction of
autoantibodies to the adrenal cortex and pancreatic islet cells by
interferon alpha therapy for chronic hepatitis C. Gut 2001;48:378-383.
[Abstract/Full Text] - Durelli L, Ferrero B, Oggero A, et al. Thyroid function and
autoimmunity during interferon beta-1b treatment: a multicenter prospective
study. J Clin Endocrinol Metab 2001;86:3525-3532.
[Abstract/Full Text] - Brix TH, Christensen K, Holm NV, Harvald B, Hegedus L. A population-based study of Graves' disease in Danish twins. Clin Endocrinol (Oxf) 1998;48:397-400. [CrossRef][ISI][Medline]
- Redondo MJ, Yu L, Hawa M, et al. Heterogeneity of type 1 diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001;44:354-362. [Erratum, Diabetologia 2001;44:927.] [CrossRef][ISI][Medline]
- Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF,
Ramsdell F. Cellular and molecular characterization of the scurfy mouse
mutant. J Immunol 1999;162:2546-2554.
[Abstract/Full Text] - Hanahan D. Peripheral-antigen-expressing cells in thymic medulla: factors in self-tolerance and autoimmunity. Curr Opin Immunol 1998;10:656-662. [CrossRef][ISI][Medline]
- Anderson MS, Venanzi ES, Klein L, et al. Projection of an
immunological self shadow within the thymus by the aire protein. Science
2002;298:1395-1401.
[Abstract/Full Text] - Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000;12:431-440. [ISI][Medline]
- Bach J-F. The effect of infections on susceptibility to
autoimmune and allergic diseases. N Engl J Med 2002;347:911-920.
[Full Text] - Ramanathan S, Bihoreau MT, Paterson AD, Marandi L, Gauguier D,
Poussier P. Thymectomy and radiation-induced type 1 diabetes in
nonlymphopenic BB rats. Diabetes 2002;51:2975-2981.
[Abstract/Full Text] - Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell 2000;101:455-458. [ISI][Medline]
- Lyons PA, Armitage N, Lord CJ, et al. Mapping by genetic
interaction: high-resolution congenic mapping of the type 1 diabetes loci
Idd10 and Idd18 in the NOD mouse. Diabetes 2001;50:2633-2637.
[Abstract/Full Text] - Robles DT, Eisenbarth GS, Dailey NJM, Peterson LB, Wicker LS. Insulin autoantibodies are associated with islet inflammation but not always related to diabetes progression in NOD congenic mice. Diabetes 2002;52:882-886. [ISI]
- Thomas HE, Kay TW. Beta cell destruction in the development of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Diabetes Metab Res Rev 2000;16:251-261. [CrossRef][ISI][Medline]
- Moriyama H, Wen L, Abiru N, et al. Induction and acceleration
of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic
acid) and an insulin self-peptide. Proc Natl Acad Sci U S A
2002;99:5539-5544.
[Abstract/Full Text] - Karges W, Pechhold K, Al Dahouk S, et al. Induction of
autoimmune diabetes through insulin (but not GAD65) DNA vaccination in
nonobese diabetic and in RIP-B7.1 mice. Diabetes 2002;51:3237-3244.
[Abstract/Full Text] - Lieberman SM, Evans AM, Han B, et al. Identification of the
beta cell antigen targeted by a prevalent population of pathogenic CD8+ T
cells in autoimmune diabetes. Proc Natl Acad Sci U S A 2003;100:8384-8388.
[Abstract/Full Text] - Hutton JC, Eisenbarth GS. A pancreatic beta-cell-specific
homolog of glucose-6-phosphatase emerges as a major target of cell-mediated
autoimmunity in diabetes. Proc Natl Acad Sci U S A 2003;100:8626-8628.
[Full Text] - Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 2000;406:739-742. [CrossRef][ISI][Medline]
- Martin AM, Maxson MN, Leif J, Mordes JP, Greiner DL, Blankenhorn EP. Diabetes-prone and diabetes-resistant BB rats share a common major diabetes susceptibility locus, iddm4: additional evidence for a "universal autoimmunity locus" on rat chromosome 4. Diabetes 1999;48:2138-2144. [Abstract]
- MacMurray AJ, Moralejo DH, Kwitek AE, et al. Lymphopenia in the
BB rat model of type 1 diabetes is due to a mutation in a novel
immune-associated nucleotide (Ian)-related gene. Genome Res
2002;12:1029-1039.
[Abstract/Full Text] - Hornum L, Romer J, Markholst H. The diabetes-prone BB rat
carries a frameshift mutation in Ian4, a positional candidate of Iddm1.
Diabetes 2002;51:1972-1979.
[Abstract/Full Text] - Awata T, Guberski DL, Like AA. Genetics of the BB rat: association of autoimmune disorders (diabetes, insulitis, and thyroiditis) with lymphopenia and major histocompatibility complex class II. Endocrinology 1995;136:5731-5735. [Abstract]
- An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 1997;17:399-403. [ISI][Medline]
- Pearce SH, Cheetham T, Imrie H, et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet 1998;63:1675-1684. [CrossRef][ISI][Medline]
- Scott HS, Heino M, Peterson P, et al. Common mutations in
autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of
different origins. Mol Endocrinol 1998;12:1112-1119.
[Abstract/Full Text] - Kumar PG, Laloraya M, She JX. Population genetics and functions of the autoimmune regulator (AIRE). Endocrinol Metab Clin North Am 2002;31:321-338. [ISI][Medline]
- H�genauer C, Meyer RL, Netto GJ, et al. Malabsorption due to
cholecystokinin deficiency in a patient with autoimmune polyglandular
syndrome type I. N Engl J Med 2001;344:270-274.
[Full Text] - Ramsey C, Winqvist O, Puhakka L, et al. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet 2002;11:397-409. [CrossRef][ISI][Medline]
- Furmaniak J, Sanders J, Rees Smith B. Autoantigens in the autoimmune endocrinopathies. In: Volp� R, ed. Autoimmune endocrinopathies. Totowa, N.J.: Humana Press, 1999:183-216.
- Muir A, Schatz DA, Maclaren NK. Polyglandular failure syndromes. In: DeGroot LJ, Besser M, Burger HG, et al., eds. Endocrinology. 3rd ed. Vol. 3. Philadelphia: W.B. Saunders, 1995:3013-24.
- Yu L, Brewer KW, Gates S, et al. DRB1*04 and DQ alleles:
expression of 21-hydroxylase autoantibodies and risk of progression to
Addison's disease. J Clin Endocrinol Metab 1999;84:328-335.
[Abstract/Full Text] - Myhre AG, Undlien DE, Lovas K, et al. Autoimmune adrenocortical
failure in Norway: autoantibodies and human leukocyte antigen class II
associations related to clinical features. J Clin Endocrinol Metab
2002;87:618-623.
[Abstract/Full Text] - Falorni A, Laureti S, Santeusanio F. Autoantibodies in autoimmune polyendocrine syndrome type II. Endocrinol Metab Clin North Am 2002;31:369-389. [ISI][Medline]
- Patel DD. Escape from tolerance in the human X-linked
autoimmunity-allergic disregulation syndrome and the Scurfy mouse. J Clin
Invest 2001;107:155-157.
[Full Text] - Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001;27:18-20. [CrossRef][ISI][Medline]
- Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and
molecular features of the immunodysregulation, polyendocrinopathy,
enteropathy, X linked (IPEX) syndrome. J Med Genet 2002;39:537-545.
[Abstract/Full Text] - Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF.
Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell
activation. J Biol Chem 2001;276:37672-37679.
[Abstract/Full Text] - Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell
development by the transcription factor Foxp3. Science 2003;299:1057-1061.
[Abstract/Full Text] - Uchigata Y, Hirata Y. Insulin autoimmune syndrome (IAS, Hirata disease). In: Eisenbarth GS, ed. Molecular mechanisms of endocrine and organ specific autoimmunity. Austin, Tex.: R.G. Landes, 1999:133-48.
- Schey S. Osteosclerotic myeloma and `POEMS' syndrome. Blood Rev 1996;10:75-80. [CrossRef][ISI][Medline]
- Rovira M, Carreras E, Blade J, et al. Dramatic improvement of POEMS syndrome following autologous haematopoietic cell transplantation. Br J Haematol 2001;115:373-375. [CrossRef][ISI][Medline]
- Hogan WJ, Lacy MQ, Wiseman GA, Fealey RD, Dispenzieri A, Gertz MA. Successful treatment of POEMS syndrome with autologous hematopoietic progenitor cell transplantation. Bone Marrow Transplant 2001;28:305-309. [CrossRef][ISI][Medline]
- Perniola R, Falorni A, Clemente MG, Forini F, Accogli E, Lobreglio G. Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Eur J Endocrinol 2000;143:497-503. [ISI][Medline]
- Falorni A, Laureti S, Nikoshkov A, et al. 21-Hydroxylase autoantibodies in adult patients with endocrine autoimmune diseases are highly specific for Addison's disease. Clin Exp Immunol 1997;107:341-346. [ISI][Medline]
- Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely
ill patients. N Engl J Med 2003;348:727-734.
[Full Text] - Avcin T, Cimaz R, Falcini F, et al. Prevalence and clinical
significance of anti-cyclic citrullinated peptide antibodies in juvenile
idiopathic arthritis. Ann Rheum Dis 2002;61:608-611.
[Abstract/Full Text] - Vincent C, Nogueira L, Sebbag M, et al. Detection of antibodies to deiminated recombinant rat filaggrin by enzyme-linked immunosorbent assay: a highly effective test for the diagnosis of rheumatoid arthritis. Arthritis Rheum 2002;46:2051-2058. [CrossRef][ISI][Medline]
- Gottlieb PA, Fain P. Genetic basis of autoimmune adrenal deficiency. Curr Opin Endocrinol Diabetes 2002;9:237-43.
- Schenker M, Hummel M, Ferber K, et al. Early expression and high prevalence of islet autoantibodies for DR3/4 heterozygous and DR4/4 homozygous offspring of parents with Type I diabetes: the German BABYDIAB study. Diabetologia 1999;42:671-677. [CrossRef][ISI][Medline]
- Halonen M, Eskelin P, Myhre AG, et al. AIRE mutations and human
leukocyte antigen genotypes as determinants of the autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy phenotype. J Clin
Endocrinol Metab 2002;87:2568-2574.
[Abstract/Full Text] - Park YS, Sanjeevi CB, Robles D, et al. Additional association of intra-MHC genes, MICA and D6S273, with Addison's disease. Tissue Antigens 2002;60:155-163. [CrossRef][ISI][Medline]
- Gambelunghe G, Falorni A, Ghaderi M, et al. Microsatellite
polymorphism of the MHC class I chain-related (MIC-A and MIC-B) genes marks
the risk for autoimmune Addison's disease. J Clin Endocrinol Metab
1999;84:3701-3707.
[Abstract/Full Text] - Kemp EH, Ajjan RA, Husebye ES, et al. A cytotoxic T lymphocyte antigen-4 (CTLA-4) gene polymorphism is associated with autoimmune Addison's disease in English patients. Clin Endocrinol (Oxf) 1998;49:609-613. [CrossRef][ISI][Medline]
- Cox NJ, Wapelhorst B, Morrison VA, et al. Seven regions of the genome show evidence of linkage to type 1 diabetes in a consensus analysis of 767 multiplex families. Am J Hum Genet 2001;69:820-830. [CrossRef][ISI][Medline]
- Alkhateeb A, Stetler GL, Old W, et al. Mapping of an
autoimmunity susceptibility locus (AIS1) to chromosome 1p31
[PDB]
.3-p32.2. Hum Mol Genet 2002;11:661-667.
[Abstract/Full Text] - Verge CF, Vardi P, Babu S, et al. Evidence for oligogenic
inheritance of type 1 diabetes in a large Bedouin Arab family. J Clin Invest
1998;102:1569-1575.
[Abstract/Full Text] - Alvarez-Marfany M, Roman SH, Drexler AJ, Robertson C, Stagnaro-Green A. Long-term prospective study of postpartum thyroid dysfunction in women with insulin dependent diabetes mellitus. J Clin Endocrinol Metab 1994;79:10-16. [Abstract]
- Coles AJ, Wing M, Smith S, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 1999;354:1691-1695. [CrossRef][ISI][Medline]
- Hoffenberg EJ, Bao F, Eisenbarth GS, et al. Transglutaminase antibodies in children with a genetic risk for celiac disease. J Pediatr 2000;137:356-360. [CrossRef][ISI][Medline]
- Shan L, Molberg O, Parrot I, et al. Structural basis for gluten
intolerance in celiac sprue. Science 2002;297:2275-2279.
[Abstract/Full Text] - Roep BO, Atkinson MA, van Endert PM, Gottlieb PA, Wilson SB, Sachs JA. Autoreactive T cell responses in insulin-dependent (Type 1) diabetes mellitus: report of the first international workshop for standardization of T cell assays. J Autoimmun 1999;13:267-282. [CrossRef][ISI][Medline]
- Nepom GT, Buckner JH, Novak EJ, et al. HLA class II tetramers: tools for direct analysis of antigen-specific CD4+ T cells. Arthritis Rheum 2002;46:5-12. [CrossRef][ISI][Medline]
- Kawasaki E, Eisenbarth GS. High-throughput radioassays for autoantibodies to recombinant autoantigens. Front Biosci 2000;5:E181-E190. [ISI][Medline]
- Yu L, Eisenbarth GS. Humoral autoimmunity. In: Eisenbarth GS, ed. Type 1 diabetes: molecular, cellular, and clinical immunology. Denver: Barbara Davis Center for Childhood Diabetes, 2002 (Web only). (Accessed March 23, 2004, at http://www.barbaradaviscenter.org.)
- Winter WE, Harris N, Schatz D. Type 1 diabetes islet autoantibody markers. Diabetes Technol Ther 2002;4:817-839. [CrossRef][Medline]
- Yu L, Robles DT, Abiru N, et al. Early expression of
antiinsulin autoantibodies of humans and the NOD mouse: evidence for early
determination of subsequent diabetes. Proc Natl Acad Sci U S A
2000;97:1701-1706.
[Abstract/Full Text] - Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996;45:926-933. [Abstract]
- Bingley PJ, Bonifacio E, Williams AJK, Genovese S, Bottazzo GF, Gale EAM. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes 1997;46:1701-1710. [Abstract]
- Achenbach P, Warncke K, Reiter J, et al. Stratification of type
1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes
2004;53:384-392.
[Abstract/Full Text] - Soderbergh A, Winqvist O, Norheim I, et al. Adrenal autoantibodies and organ-specific autoimmunity in patients with Addison's disease. Clin Endocrinol (Oxf) 1996;45:453-460. [ISI][Medline]
- Bao F, Yu L, Babu S, et al. One third of HLA DQ2 homozygous patients with type 1 diabetes express celiac disease associated transglutaminase autoantibodies. J Autoimmun 1999;13:143-148. [CrossRef][ISI][Medline]
- Chase HP, Cuthbertson DD, Dolan LM, et al. First-phase insulin release during the intravenous glucose tolerance test as a risk factor for type 1 diabetes. J Pediatr 2001;138:244-249. [CrossRef][ISI][Medline]
- De Block CE, De Leeuw IH, Decochez K, et al. The presence of
thyrogastric antibodies in first degree relatives of type 1 diabetic
patients is associated with age and proband antibody status. J Clin
Endocrinol Metab 2001;86:4358-4363.
[Abstract/Full Text] - Corazza GR, Di Sario A, Cecchetti L, et al. Influence of pattern of clinical presentation and of gluten-free diet on bone mass and metabolism in adult coeliac disease. Bone 1996;18:525-530. [CrossRef][ISI][Medline]
- Major cross-country differences in risk of dying for people with IDDM. Diabetes Care 1991;14:49-54. [Abstract]
- Sartor G, Dahlquist G. Short-term mortality in childhood onset insulin-dependent diabetes mellitus: a high frequency of unexpected deaths in bed. Diabet Med 1995;12:607-611. [ISI][Medline]
- Diabetes Prevention Trial-Type 1 Diabetes Study Group. Effects
of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J
Med 2002;346:1685-1691.
[Abstract/Full Text] - Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal
antibody in new-onset type 1 diabetes mellitus. N Engl J Med
2002;346:1692-1698.
[Abstract/Full Text] - Alleva DG, Gaur A, Jin L, et al. Immunological characterization
and therapeutic activity of an altered-peptide ligand, NBI-6024, based on
the immunodominant type 1 diabetes autoantigen insulin B-chain (9-23)
peptide. Diabetes 2002;51:2126-2134.
[Abstract/Full Text] - Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med
2002;346:180-188.
[Full Text] - Collin P, Kaukinen K, Valimaki M, Salmi J. Endocrinological
disorders and celiac disease. Endocr Rev 2002;23:464-483.
[Abstract/Full Text] - Sutherland DE, Sibley R, Xu XZ, et al. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans Assoc Am Physicians 1984;97:80-87. [ISI][Medline]
- Venstrom JM, McBride MA, Rother KI, Hirshberg B, Orchard TJ,
Harlan DM. Survival after pancreas transplantation in patients with diabetes
and preserved kidney function. JAMA 2003;290:2817-2823.
[Abstract/Full Text] - Ryan EA, Lakey JR, Paty BW, et al. Successful islet
transplantation: continued insulin reserve provides long-term glycemic
control. Diabetes 2002;51:2148-2157.
[Abstract/Full Text] - Bosi E, Braghi S, Maffi P, et al. Autoantibody response to
islet transplantation in type 1 diabetes. Diabetes 2001;50:2464-2471.
[Abstract/Full Text]