UK
Europe
USA&Canada
Aust.&NZ
Asia
UK
Europe
USA&Canada
Aust.&NZ
Asia
Editorials Online�
Medical
Law News
E-texts
Links
Classifieds
Home
|
|
|
NEJM
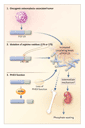
Josef T. Prchal, M.D. The discovery of the DNA code touched off an explosion of our knowledge of the molecular basis of human diseases and sparked ideas about treating inherited diseases with genes. Unfortunately, the first widely publicized attempt at gene therapy, in which the -globin gene was injected into a patient with -thalassemia, undertaken 20 years ago, failed because it was not based on an understanding of gene regulation. This ill-conceived experiment led to a public backlash and an unwarranted fear, bordering on hysteria, of recombinant-DNA research and its clinical applications. It soon became clear that identifying a gene sequence and a disease-causing mutation was not a sufficient basis for gene therapy. Understanding gene regulation, including tissue-specific control of gene expression, and developing an appropriate strategy to correct the defect involved in the disease (e.g., increasing production of factor VIII in hemophilia or producing an antisickling globin in sickle cell disease), are mandatory for successful gene therapy. It is also important to deliver and express the disease-correcting gene and to maintain its activity. Substantial progress has been made in overcoming these obstacles, as demonstrated by recent reports of the use of lentivirus vectors to cure thalassemia and sickle cell disease in mice. Although gene therapy in mice has often been plagued by overexpression of the inserted gene, the main problem in humans is low levels of expression of the new gene. Even so, a very low level of expression of the gene may be sufficient for a clinically beneficial effect in hemophilia. Binley et al. have made an important advance in gene therapy1 by showing that erythropoietin can be produced on demand in erythropoietin-deficient mice. Treatment with recombinant erythropoietin, although costly, has improved the quality of life of innumerable patients who have anemia as a result of renal disease and other causes. The work of Binley et al. suggests that an elegant gene-therapy maneuver could regulate the delivery of erythropoietin. Their work exploits the rapidly expanding understanding of the regulation of genes by hypoxia, including those involved in crucial processes such as embryogenesis, vasculogenesis, and energy metabolism. Erythropoietin is the first and the best-studied example of a gene that is regulated by the hypoxia sensor pathway. The 3'-flanking region of the erythropoietin gene contains a hypoxia response element, which is required to induce transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1), a unique transcription factor that binds to the hypoxia response element, is ubiquitous and functions as a master regulator of many genes that are responsive to hypoxia.2 The hypoxia sensor (Figure 1) is a complex containing the oxygen-sensitive HIF-1 subunit, which is rapidly destroyed in the presence of oxygen.3 In a reaction requiring oxygen and iron, a novel proline hydroxylase hydroxylates HIF-1 and renders it susceptible to ubiquitination by the von Hippel�Lindau protein and to subsequent degradation in the proteosome. In the absence of oxygen, HIF-1 cannot be degraded (Figure 1); thus, there is a rapid increase in HIF-1 and up-regulation of hypoxia-responsive genes, including erythropoietin. One of the key components of this process, von Hippel�Lindau protein, is mutated in a cancer-predisposition syndrome inherited in an autosomal dominant fashion that includes renal-cell carcinoma, pheochromocytoma, and cerebral hemangioma. Curiously, cerebral hemangiomas are often associated with polycythemia as a result of high erythropoietin levels. Recently, a congenital disorder of hypoxia sensing that was due to a homozygous germ-line mutation in the von Hippel�Lindau gene4 was found to cause polycythemia with increased erythropoietin levels but not an increased susceptibility to cancer.
Knowledge of hypoxia sensing was essential for the success of Binley and coworkers. In their gene-therapy experiment, they placed the erythropoietin gene under the control of the hypoxia response element, which conferred hypoxic responsiveness on the gene product. In this way, erythropoietin was produced in response to the hypoxia caused by anemia. Two other factors were important: muscles injected with DNA efficiently produce the corresponding protein, and hypoxia-sensing mechanisms function ubiquitously in mammals. Binley et al. demonstrated that the muscle of anemic mice was hypoxic and allowed the production of erythropoietin, which ceased with the correction of anemia. In contrast, control mice injected with an erythropoietin gene driven by a continously acting promoter that was not sensitive to hypoxia became polycythemic. Clearly, these findings represent progress, but more work needs to be done before treatment with recombinant erythropoietin can be replaced by a single injection of DNA, which would last a lifetime. Most likely, oxygen demand, which is constant in the kidney, varies in muscle because of exercise-induced hypoxia. Furthermore, there may be differences in the degree of hypoxia sensing in different tissues; several genes encode the proline hydroxylases, and little is known about their distribution in tissue. Indeed, careful examination of the mice treated with the erythropoietin gene controlled by the hypoxia response element reveals a wide range of hematocrit values from anemia to mild polycythemia, although not as extreme as the polycythemia in normal mice injected with the vector without the hypoxia response element. More data on the physiological regulation of erythropoietin expression in the treated mice are needed to justify the conclusion of Binley et al. that erythropoietin secretion is "physiologically regulated." There is also concern that tissue that is not normally programmed to synthesize erythropoietin might produce a protein with conformational changes that would lead to an immune response, as has been encountered in rare patients who have received recombinant erythropoietin. Nevertheless, the work of Binley et al. is a step in the right direction.
Supported by grants (R01HL66333-02 and R01HL5007-09) from the
National Heart, Lung, and Blood Institute, National Institutes of
Health, Bethesda, Md.
From the Department of Medicine, Baylor College of Medicine, Houston. References
|