|
NEJM
|
Robert S. Schwartz, M.D.
Patients with sustained high-grade eosinophilia without an evident cause usually receive the diagnosis of hypereosinophilic syndrome, a high-sounding term that only masks our ignorance. Apart from the consistent presence of hypereosinophilia, the clinical picture of the hypereosinophilic syndrome varies. In some cases, the toxic contents of tissue-invasive eosinophils (see electron micrograph) cause heart failure, pulmonary lesions, and harm to other organs. In other cases, urticaria and itchy nodules in the skin dominate the clinical picture. The hypereosinophilic syndrome is a rare disorder, yet as Cools et al. tell us in this issue of the Journal (pages 1201�1214), it carries a message of general interest concerning the fundamental biology of cancer: chromosomal aberrations that can be detected by standard karyotyping, a common feature of many kinds of neoplasms, is a marker for a cryptic molecular arrangement that has created an oncogenic partnership between two otherwise innocent genes.
|

The production of eosinophils requires three cytokines: interleukin-5, interleukin-3, and granulocyte�macrophage colony-stimulating factor. Cells destined for the eosinophilic lineage display high-affinity receptors for interleukin-5, a specific differentiation factor for eosinophils. In rare instances, lymphoma or leukemia cells overproduce interleukin-5, thereby evoking hypereosinophilia. In most cases of idiopathic hypereosinophilic syndrome, however, the eosinophils are independent of growth factors, and the condition has features of a myeloproliferative syndrome: hepatosplenomegaly, thrombocytopenia, abnormal chromosomes, and clonal populations of eosinophils.
In another type of hypereosinophilic syndrome, a monoclonal population of T cells lurks behind the eosinophils. These T cells are activated, display abnormal combinations of surface markers, and produce large amounts of interleukin-5, the presumed cause of the eosinophilia. IgE levels in serum can be very high, perhaps because of other cytokines released by the T cells. Many patients with this variant of the hypereosinophilic syndrome have an intractable dermatitis. In these cases, the peculiar T cells, especially those with abnormal chromosomes, seem to be headed toward neoplastic transformation. Indeed, biopsies of the dermatitis may show signs suggestive of cutaneous T-cell lymphoma, and frank lymphoma can develop in the lymph nodes.
Recently, several groups have reported that some cases of the hypereosinophilic syndrome respond to imatinib mesylate. This is good news, because the syndrome is inexorable and often fatal. Imatinib was originally used to treat chronic myelogenous leukemia, which is caused by a cytogenetic defect termed t(9;22)(q34;q11), in which breaks form in chromosomes 9 and 22, which then exchange chromosomal fragments. This reciprocal translocation ("t"), in which the breaks occur at band q34 on the long arm ("q") of chromosome 9 and at band q11 of chromosome 22, creates strange genetic bedfellows. It fuses pieces of DNA from two genes, BCR and ABL, forming a new gene, BCR-ABL. The product of the conjoined genes, the BCR-ABL fusion protein, is a ceaselessly active enzyme (a tyrosine kinase) that is sensitive to suppression by imatinib, a specific inhibitor of several tyrosine kinases. Since the BCR-ABL translocation does not occur in the hypereosinophilic syndrome, the recent results with imatinib point to a molecular lesion in the disease that results in a new kind of tyrosine kinase. Cools et al. confirm these clinical reports and reveal the nature of the lesion.
Of the 11 patients Cools et al. treated with imatinib, 10 entered complete remission (1 of them only transiently). All of them had life-threatening manifestations of the hypereosinophilic syndrome. The key that unlocked the secret to the molecular lesion was held by a patient with a translocation termed t(1;4)(q44;q12). The villain emerged when Cools et al. found that a deletion of chromosomal material from 4q12 had left behind fragments of two genes, FIP1L1 and PDGFRA, which fused to form a novel gene, FIP1L1-PDGFRA, as shown in the diagram (see Figure). By an extraordinary coincidence, the product of PDGFRA, PDGFR, is a tyrosine kinase, and its partner in the fusion protein, FIP1L1, keeps PDGFR continuously active. Now we can understand the clinical responses of the hypereosinophilic syndrome to imatinib, because the constitutively active FIP1L1-PDGFR tyrosine kinase is analogous to the imatinib-sensitive BCR-ABL enzyme.
|
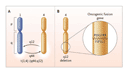
The FIP1L1-PDGFR fusion protein is a plausible cause of some cases of the hypereosinophilic syndrome, especially in view of its oncogenic properties in vitro. Other molecular abnormalities are likely to be found, however, because a search for FIP1L1-PDGFRA in 16 patients with the hypereosinophilic syndrome uncovered it in only 9. This is not a surprising result, given the heterogeneity of the syndrome. Interestingly, of the four patients with dermatologic manifestations, two had the FIP1L1-PDGFRA abnormality. Whether any of these patients with skin lesions had a monoclonal population of T cells is unknown.
Also notable is the previously reported fusion gene involving PDGFRB (EVT6-PDGFRB) that has been found in patients with certain chronic myeloproliferative diseases with high-grade eosinophilia. PDGFR and the PDGFR involved in the hypereosinophilic syndrome are receptors for members of the platelet-derived growth factor (PDGF) family. They are tyrosine kinases, and when ligated by the corresponding growth factor, they signal cells to proliferate. The EVT6-PDGFR fusion protein is a constantly active tyrosine kinase with oncogenic properties and is inhibited by imatinib. What remains to be solved is the problem of how these growth factor receptors relate to hypereosinophilia.
The discovery of FIP1L1-PDGFRA owes a considerable debt to the simple observation that phytohemagglutinin, a plant lectin, triggers the division of mammalian cells in vitro, thus allowing easy visualization of chromosomes. In 1960, Nowell and Hungerford1 used this phenomenon to discover the Philadelphia chromosome of chronic myelogenous leukemia. The subsequent finding that the "minute chromosome" they described is a reciprocal translocation that results in a leukemogenic protein, BCR-ABL, laid the foundation for innumerable advances in the molecular genetics of neoplastic diseases, of which the discovery of FIP1L1-PDGFRA is an example. As we prepare to celebrate the 50th anniversary of the discovery of the double-helical structure of DNA, it is clear that the union of molecular biology with clinical medicine, exemplified by the article by Cools et al., is now paying handsome dividends in the clinic.
References
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960;132:1497-1497. [ISI]