NEJM
Review Article
|
Bruce C. Trapnell, M.D., Jeffrey A. Whitsett, M.D., and Koh Nakata, M.D., Ph.D.
Pulmonary alveolar proteinosis is a rare disorder in which lipoproteinaceous material accumulates within alveoli.1 The clinical course of the disease is variable, ranging from respiratory failure to spontaneous resolution. An important feature of the disease is susceptibility to pulmonary infections, sometimes with opportunistic organisms.
Pulmonary alveolar proteinosis occurs in three clinically distinct forms:
congenital, secondary, and acquired. The congenital form comprises a
heterogeneous group of disorders2 caused by
mutations in the genes encoding surfactant protein B or C or the ![]() C
chain of the receptor for granulocyte�macrophage colony-stimulating
factor (GM-CSF).3,4,5,6,7
Secondary pulmonary alveolar proteinosis develops in association with
conditions involving functional impairment or reduced numbers of
alveolar macrophages. Such conditions include some hematologic
cancers, pharmacologic immunosuppression, inhalation of inorganic
dust (e.g., silica) or toxic fumes, and certain infections.8,9,10,11,12,13,14,15
Acquired (or idiopathic) pulmonary alveolar proteinosis has been an
enigmatic and fascinating disorder since its initial description, in
1958.1 Recent observations in transgenic
mice and humans, however, have provided important clues to its
pathogenesis. In this review, we highlight the ways in which these
studies led to the concept that acquired pulmonary alveolar
proteinosis is an autoimmune disease targeting GM-CSF and the ways in
which the critical role of GM-CSF in the lung was identified.
C
chain of the receptor for granulocyte�macrophage colony-stimulating
factor (GM-CSF).3,4,5,6,7
Secondary pulmonary alveolar proteinosis develops in association with
conditions involving functional impairment or reduced numbers of
alveolar macrophages. Such conditions include some hematologic
cancers, pharmacologic immunosuppression, inhalation of inorganic
dust (e.g., silica) or toxic fumes, and certain infections.8,9,10,11,12,13,14,15
Acquired (or idiopathic) pulmonary alveolar proteinosis has been an
enigmatic and fascinating disorder since its initial description, in
1958.1 Recent observations in transgenic
mice and humans, however, have provided important clues to its
pathogenesis. In this review, we highlight the ways in which these
studies led to the concept that acquired pulmonary alveolar
proteinosis is an autoimmune disease targeting GM-CSF and the ways in
which the critical role of GM-CSF in the lung was identified.
Epidemiology
The prevalence of acquired pulmonary alveolar proteinosis has been estimated to be 0.37 per 100,000 persons.16 It is a primary acquired disorder in more than 90 percent of cases.7,17,18,19 The median age at the time of diagnosis is 39 years; most patients are men, and 72 percent have a history of smoking.19 The male predominance may be linked to the more frequent use, historically, of tobacco by men.19
Clinical, Radiographic, and Laboratory Manifestations
Clinical Presentation
Most patients with acquired pulmonary alveolar proteinosis present with progressive exertional dyspnea of insidious onset and cough.1,17,18,19,20 Less commonly, fever, chest pain, or hemoptysis also occurs, especially if secondary infection is present. The history does not include clinically significant environmental pulmonary exposures or other potential causes. The findings on physical examination can be unremarkable, but there are inspiratory crackles in 50 percent of patients, cyanosis in 25 percent, and digital clubbing in a small percentage. Several reviews,17,18,19 including an excellent analysis of data from 410 patients, accounting for most if not all of the published cases,19 provide further details on the clinical presentation, demographics, and clinical course of patients with acquired pulmonary alveolar proteinosis.
In uncomplicated pulmonary alveolar proteinosis, the chest radiograph usually reveals bilateral air-space disease with an ill-defined nodular or confluent pattern, often with a perihilar predominance suggestive of the "bat wing" appearance of pulmonary edema but without other radiographic signs of left-sided heart failure (Figure 1A).1,21,22 Notably, the extent of radiographic abnormalities is often disproportionately increased relative to the severity of the symptoms and physical findings. High-resolution computed tomography shows patchy, ground-glass opacifications with superimposed interlobular septal and intralobular thickening, a pattern commonly referred to as "crazy paving" (Figure 1B).23,24 Though not specific for pulmonary alveolar proteinosis,24 the extent and severity of these radiographic findings correlate with the degree of impairment in pulmonary function as measured by spirometry or arterial blood gas analysis.23
|

Laboratory Findings
In acquired pulmonary alveolar proteinosis, routine blood counts and the results of routine blood chemical analysis and urinalysis are usually normal.18,22,25 The serum level of lactate dehydrogenase is frequently slightly elevated26 and may be a useful marker of the severity of the disease.19,26 Elevations in the serum levels of carcinoembryonic antigen,27 cytokeratin 19,28 mucin KL-6,29 and surfactant proteins A, B, and D30,31 are of unclear prognostic value.
Pulmonary Function
The results of tests of pulmonary function can be normal, but typically they show a restrictive ventilatory defect with slight impairments in the forced vital capacity and total lung capacity and a disproportionate, severe reduction of the carbon monoxide diffusing capacity.19,32 Hypoxemia is caused by ventilation�perfusion inequality and intrapulmonary shunting, resulting in a widened alveolar�arteriolar diffusion gradient.19,33
Characteristics of Bronchoalveolar-Lavage Fluid
Clinical and radiographic findings often suggest the diagnosis of pulmonary alveolar proteinosis1,22,34; in about 75 percent of suspected cases, findings on examination of a bronchoalveolar-lavage specimen can establish the diagnosis.22 The lavage fluid in patients with this disorder has an opaque, milky appearance (Figure 2A). It contains large and foamy alveolar macrophages (Figure 2B) or monocyte-like alveolar macrophages and increased numbers of lymphocytes35,36 but relatively few inflammatory cells of other types. There are also large, acellular, eosinophilic bodies in a diffuse background of granular material that stains with periodic acid�Schiff, as well as elevated levels of surfactant proteins.22,36 Electron microscopy shows that the intraalveolar material consists of amorphous, granular debris containing numerous osmiophilic, fused membrane structures with a periodicity of 4.7 nm and resembling lamellar bodies and tubular myelin (Figure 2C).
|
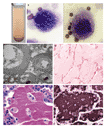
Pathological Features
Open-lung biopsy is the gold standard for the diagnosis of pulmonary alveolar proteinosis, but it is not always required and can be complicated by false negative results due to sampling error.1,22,37 On light-microscopical examination, the architecture of the lung parenchyma is preserved unless there is infection. The walls of transitional airways and alveoli are usually normal (Figure 2D), but sometimes they are thickened by lymphocytic infiltration or, less commonly, fibrosis. Alveoli are filled with granular, eosinophilic material that stains with periodic acid�Schiff (Figure 2D and Figure 2E) and within which intact and degenerating macrophages are usually evident. Immunohistochemical staining reveals abundant accumulation of surfactant protein (Figure 2F). A useful serologic test for the disease (discussed below) has been developed.38
Natural History
In any given case of acquired pulmonary alveolar proteinosis, the clinical course falls into one of three categories: stable but with persistent symptoms, progressive deterioration, or spontaneous improvement.1 A retrospective analysis of 303 cases19 found clinically significant spontaneous improvement in 24 (8 percent). In a retrospective analysis of 343 cases, the five-year survival rate was about 75 percent.19 Of the deaths in that study, 72 percent were directly due to respiratory failure from pulmonary alveolar proteinosis and 20 percent were due to pulmonary alveolar proteinosis with uncontrolled infection.
Patients with acquired pulmonary alveolar proteinosis are at risk for infections from a variety of pathogens.18,19,39 Although such infectious agents include common respiratory pathogens, opportunistic pathogens (especially nocardia) are common.18,19,40 Interestingly, infections in pulmonary alveolar proteinosis frequently occur at sites outside the lung, suggesting systemic defects in host defense.19,41,42,43
Surfactant Homeostasis
Surfactant plays a vital part in reducing surface tension at the air�liquid interface of the alveolar wall, thus preventing alveolar collapse and transudation of capillary fluid into the alveolar lumen.44 About 90 percent of surfactant is lipid (predominantly phospholipid), 10 percent is protein, and less than 1 percent is carbohydrate. Surfactant proteins A, B, C, and D contribute to the surface-active properties and structural forms of intraalveolar surfactant,45 participate in surfactant metabolism,46 opsonize microbial pathogens,47 and stimulate the defensive functions of alveolar macrophages.48 Surfactant lipids and proteins are synthesized, stored, and secreted into the alveoli by alveolar type II epithelial cells and are cleared by uptake into alveolar type II cells and alveolar macrophages (Figure 3A). The size of the surfactant pool is tightly regulated by mechanisms controlling the synthesis, recycling, and catabolism of surfactant.49
|
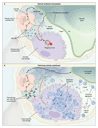
In their initial description of pulmonary alveolar proteinosis,1 Rosen et al. established that the eosinophilic material within the alveoli was rich in lipids and that it contained proteins and carbohydrates. Similarities between this material1 and the substance lining the normal alveolar wall50,51 suggested an abnormality in the production, degradation, or structure of this surface-active material52 in the disorder.53 These similarities and the identification of defects in the clearance, but not the synthesis, of alveolar phospholipid pointed to an underlying defect in the clearance of surfactant.54 The results of ultrastructural,37,55 biochemical,56,57 and functional58 investigations, together with the results of studies in genetically modified mice (discussed below), strongly support the concept that the alveolar material in pulmonary alveolar proteinosis is in fact surfactant, which accumulates due to reduced clearance rather than to overproduction.59
Mouse Models
An important clue to the pathogenesis of pulmonary alveolar proteinosis came in 1994, with the discovery that a pulmonary disorder similar to the acquired form of the disease in humans developed in knockout mice that were deficient in GM-CSF.60,61 GM-CSF, a 23-kD hematologic growth factor,62 is encoded by a gene the structure and pattern of expression of which are similar in humans and mice.63,64 The biologic effects of GM-CSF are initiated when it binds to cell-surface receptors on various hematopoietic cells, including monocytes and macrophages, and other cells, including alveolar type II epithelial cells.65,66,67,68,69 Until 1994, the principal biologic effects of GM-CSF were thought to be stimulation of the production of myeloid cells from hematopoietic precursors and enhancement of some immune functions in mature myeloid cells.64,70 Indeed, GM-CSF is used to ameliorate chemotherapy-induced neutropenia and to hasten hematopoietic recovery after bone marrow transplantation.67,71
GM-CSF and Surfactant Homeostasis
Targeted disruption of the gene encoding GM-CSF or the gene encoding
the ![]() C chain of the GM-CSF
receptor in mice (GM�/� and
C chain of the GM-CSF
receptor in mice (GM�/� and ![]() C�/�
mice, respectively) causes accumulations of eosinophilic
lipoproteinaceous material and large, foamy macrophages in the
alveoli.60,61,72,73
The alveolar material contains tubular myelin and lamellar bodies as
well as surfactant phospholipids and surfactant proteins at
dramatically increased levels.74 Except
for a reduction in the number of eosinophils in the blood, these mice
had no base-line hematologic abnormalities.
C�/�
mice, respectively) causes accumulations of eosinophilic
lipoproteinaceous material and large, foamy macrophages in the
alveoli.60,61,72,73
The alveolar material contains tubular myelin and lamellar bodies as
well as surfactant phospholipids and surfactant proteins at
dramatically increased levels.74 Except
for a reduction in the number of eosinophils in the blood, these mice
had no base-line hematologic abnormalities.
Studies of the lungs of GM�/� mice disclosed that levels of messenger RNA for surfactant proteins A, B, and C were not altered relative to those in control mice, suggesting that the biosynthesis of these proteins was not increased.60 The secretion of surfactant phospholipids into the alveolar space also was not increased, but pulmonary phospholipid clearance was severely impaired, resulting in an increase in the size of the alveolar phospholipid pool by a factor of 6.3.74 Pulmonary clearance of surfactant protein A was also impaired. The abnormal accumulation of surfactant phospholipids and proteins in pulmonary alveolar proteinosis in both humans and mice suggested that there was a defect in the catabolism of surfactant by alveolar macrophages. This hypothesis was supported by findings on examination of alveolar macrophages recovered from GM�/� mice. Despite increased uptake by the alveolar macrophages of surfactant phospholipids and proteins, the catabolism of these molecules was severely impaired.75
Effect of GM-CSF Replacement
The efficacy of GM-CSF replacement was assessed in GM�/� mice by three methods: administration of GM-CSF,76 expression of the GM-CSF gene in the lungs of double-transgenic mice with the use of a lung-specific promoter from the gene encoding surfactant protein C (SPC-GM+/+/GM�/� mice),77 and expression of GM-CSF in the lungs of GM�/� mice after adenovirus-mediated transfer of the GM-CSF gene.78 Each of these distinct approaches resulted in resolution of the pulmonary alveolar proteinosis. The site of action of GM-CSF must have been within the lung, because the GM-CSF levels were high in the lungs but undetectable in the blood of SPC-GM+/+/GM�/� mice.77 Moreover, pulmonary, but not systemic, administration of GM-CSF resulted in resolution of the disorder in GM�/� mice.76
Cellular Target of GM-CSF
Notwithstanding, these studies did not identify the cellular target
of GM-CSF: was it the alveolar macrophage or the alveolar type II
epithelial cell? This question was answered during studies in the ![]() c�/�
mouse, in which both cell types are unresponsive to GM-CSF because of
the absence of the high-affinity GM-CSF receptor.66,73
Transplantation of bone marrow from normal mice corrected the
defective metabolism of surfactant in the
c�/�
mouse, in which both cell types are unresponsive to GM-CSF because of
the absence of the high-affinity GM-CSF receptor.66,73
Transplantation of bone marrow from normal mice corrected the
defective metabolism of surfactant in the ![]() c�/�
mice.72 Since the alveolar macrophages, but not
the alveolar type II epithelial cells, in the recipient mice were of
donor origin, we can conclude that bone marrow�derived alveolar
macrophages are the principal target of GM-CSF replacement.72
c�/�
mice.72 Since the alveolar macrophages, but not
the alveolar type II epithelial cells, in the recipient mice were of
donor origin, we can conclude that bone marrow�derived alveolar
macrophages are the principal target of GM-CSF replacement.72
Immune Functions of Alveolar Macrophages and GM-CSF
Prompted by the high risk of infections in acquired pulmonary alveolar proteinosis, investigators examined host defenses in GM�/� mice. These mice are susceptible to pulmonary infection by group B streptococcus79 and Pneumocystis carinii (after CD4+ depletion)80 and have severely impaired pulmonary clearance of bacterial, fungal, and viral pathogens.79,80,81 Of note, primary and cultured alveolar macrophages from GM�/� mice have defects in cellular adhesion, expression of pathogen-recognition receptors, phagocytosis, superoxide production, microbial killing, and secretion of proinflammatory cytokines.79,80,81,82,83,84 All these abnormalities were corrected by restoring pulmonary expression of GM-CSF. Hence, it could be concluded that this factor has a critical role in protecting the lung against infection and that it carries out this role by acting locally, within the lung itself.
Role of the Transcription Factor PU.1
The diversity of the abnormalities in alveolar macrophages in GM�/� mice suggested that the maturation of these macrophages was defective. Indeed, pulmonary GM-CSF stimulates the production of high levels of PU.1 in alveolar macrophages.84 PU.1 is a transcription factor that promotes the growth and differentiation of myeloid progenitors and that is required for the production of macrophages.85,86,87,88,89,90,91,92 Transfection of the PU.1 gene into cultured alveolar macrophages from GM�/� mice corrected all the alveolar-macrophage abnormalities described above and, it is important to note, also corrected abnormalities in the catabolism of surfactant lipids and protein59,81,82,84 (Figure 4).
|

Lessons from Animal Models
Thus, studies in mouse models of pulmonary alveolar proteinosis revealed the critical roles of GM-CSF in surfactant homeostasis and in alveolar-macrophage�mediated protection of the lung against infection. GM-CSF acts within the lung by stimulating the terminal differentiation of alveolar macrophages, principally by raising the levels of PU.1. The accumulation of alveolar surfactant in GM�/� mice is due to a defect in surfactant clearance by alveolar macrophages, and not to an increase in production.
Pathogenesis in Humans
Initially, it was thought that an inhaled irritant (e.g., silica) or infectious agent that increased the production of the natural material lining the alveoli caused pulmonary alveolar proteinosis.25,53 However, the inability to find such agents in the lung-biopsy specimens of most patients with the disorder failed to support this idea. The strong association between acquired pulmonary alveolar proteinosis and smoking suggests that there is a link between the two, but nothing more is known about this association.
Role of Autoimmunity
Inhibition of Alveolar Macrophages
The alveolar macrophages in acquired pulmonary alveolar proteinosis contain giant secondary lysosomes filled with the same material that accumulates within the alveoli,93 and they have defects in chemotaxis,93 adhesion,93 phagocytosis,94 microbicidal activity,93 and phagolysosome fusion.95 This puzzling array of abnormalities was initially attributed to excessive ingestion of lipoproteinaceous material.96 However, that idea was difficult to reconcile with the discovery of a substance, found in bronchoalveolar-lavage fluid from patients with pulmonary alveolar proteinosis, that caused normal alveolar macrophages to acquire some of those abnormalities.97,98 Furthermore, a factor found in both pulmonary-lavage fluid and serum from patients with pulmonary alveolar proteinosis blocked mitogen-stimulated proliferation of normal monocytes, suggesting the involvement of a circulating inhibitor in the pathogenesis of the disease.99
Role of GM-CSF
The demonstration that GM-CSF deficiency caused pulmonary alveolar proteinosis in mice prompted a reevaluation of the pathogenesis of the acquired form of the disease in humans. A clue came from a report that the systemic administration of recombinant human GM-CSF had produced radiographic, physiological, and symptomatic improvement in one affected patient.100 Similar treatment of additional patients (discussed below) failed to produce the expected neutrophilia100 � a curious finding that was confirmed in subsequent studies.30,101 Attempts to identify mutations in the genes encoding GM-CSF and its receptor in acquired pulmonary alveolar proteinosis have been unsuccessful to date,102 in contrast to findings in the congenital form of the disease.15 Furthermore, the levels of GM-CSF in bronchoalveolar-lavage fluid and plasma are actually elevated in the acquired form, thus ruling out the possibility that the disease is due to the absence of GM-CSF itself.103
Autoantibodies against GM-CSF
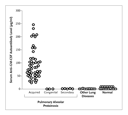
An immunologic explanation for these observations was revealed by a reexamination of the inhibitory factor in pulmonary alveolar proteinosis. Bronchoalveolar-lavage fluid from subjects with the disease, but not that from control subjects, inhibited the ability of GM-CSF to stimulate the proliferation of normal monocytes and a GM-CSF�dependent cell line and competitively inhibited the binding of GM-CSF to cells bearing GM-CSF receptors.104 This inhibitory activity was due to a neutralizing IgG antibody against GM-CSF.105 The antibody was present in bronchoalveolar-lavage fluid and serum from all patients with acquired pulmonary alveolar proteinosis but not those with the congenital or secondary form of the disorder, those with several other lung disorders, or normal controls (Figure 5).105,106 The specific association between neutralizing anti�GM-CSF autoantibodies and acquired pulmonary alveolar proteinosis38,105,106 strongly supports the view that in this disorder, a neutralizing autoantibody against GM-CSF causes defects in the functioning of alveolar macrophages, including impairment of the catabolism of surfactant lipids and proteins and disruption of surfactant homeostasis. Further strong support for this concept comes from the recent demonstration that the presence of these antibodies is correlated with the elimination of GM-CSF bioactivity in the lungs of patients with pulmonary alveolar proteinosis.107 The finding of this autoantibody has led to the development of a latex-agglutination test with high sensitivity (100 percent) and specificity (98 percent) for diagnosing the acquired disease.38
|
Pulmonary Cytokines
Similarities between pulmonary alveolar proteinosis in mice and the acquired form of the disease in humans also include abnormalities of pulmonary cytokines. For example, the level of macrophage colony-stimulating factor, which is elevated in the lungs of GM�/� mice,84 is also elevated in the lungs of humans with acquired pulmonary alveolar proteinosis.106 Similarly, the level of monocyte chemotactic protein 1 is elevated in the lungs of both GM�/� mice and humans with the acquired disease.35,83 The mechanism of these cytokine changes is not known, but the latter may explain the increased numbers of lymphocytes in the lungs of mice60 and patients with pulmonary alveolar proteinosis.35
Therapeutic Approaches
Current Approaches
The treatment of pulmonary alveolar proteinosis depends on the underlying cause. Current therapy for the congenital form of the disorder is supportive,7 although successful lung transplantation has been reported.108 Therapy for secondary pulmonary alveolar proteinosis generally involves treatment of the underlying condition; for example, when the disorder is associated with a hematologic cancer, successful chemotherapy or bone marrow transplantation corrects the associated pulmonary disorder.12
Acquired pulmonary alveolar proteinosis has been treated successfully since the early 1960s by whole-lung lavage, and this procedure remains the standard of care today.109,110,111,112,113 Although it has not been tested in prospective, randomized trials, whole-lung lavage improves clinical, physiological, and radiographic findings. A retrospective analysis of 231 cases found clinically significant improvement in arterial oxygen tension and in measures of pulmonary function (forced expiratory volume in one second, vital capacity, and diffusing capacity for carbon monoxide).19 Such therapy also improves survival: in a group of 146 patients, the mean (�SD) rate of survival at five years was 94�2 percent with lavage, as compared with 85�5 percent without lavage (P=0.04).19 The median duration of clinical benefit from lavage has been reported to be 15 months.19 Interestingly, therapeutic whole-lung lavage improves defects in the migration114 and phagocytosis115 of alveolar macrophages. Successful treatment of pulmonary alveolar proteinosis by lobar lavage through fiberoptic bronchoscopy has also been reported, although the practical clinical utility of this approach is unclear.116
GM-CSF Therapy
Several prospective phase 2 trials of GM-CSF therapy for acquired pulmonary alveolar proteinosis have been undertaken. The first, conducted from 1995 through 1998, evaluated the effectiveness of subcutaneous GM-CSF (at a dose of 5 �g per kilogram of body weight per day) for 6 to 12 weeks in 14 patients.30 Five patients had a response to this dose, with a mean improvement in the alveolar�arteriolar diffusion gradient of 23.2 mm Hg; four of the patients who did not have a response then received 20 �g per kilogram per day and had a response to that dose. The remaining five patients did not have a response at the higher dose. An ongoing study, initiated in 1998, reported a response in three of four initial patients who received daily subcutaneous injections of GM-CSF in escalating doses over a period of 12 weeks.101,117 These three patients had symptomatic, physiological, and radiographic improvement as well as a reduction in the mean alveolar�arteriolar diffusion gradient from 48.3 mm Hg at base line to 18.3 mm Hg after 16 weeks of treatment. These initial results are encouraging, but the mechanism of the effect of GM-CSF treatment is unclear. The observation of a reduction in pulmonary levels of anti�GM-CSF antibody in association with clinical improvement suggests that desensitization to GM-CSF may be involved.36
Conclusions
Clinical investigations, research in transgenic mice, and translation of findings from the bench to the bedside have considerably changed our concepts of the pathogenesis and treatment of pulmonary alveolar proteinosis. In addition to illuminating the mechanism of this disorder, research has revealed critical roles for GM-CSF in the regulation of mature alveolar macrophages in the lung, the regulation of surfactant homeostasis, and the stimulation of multiple mechanisms that protect the lung against microbial invasion.
Supported in part by grants (HL69549 and HL71823, to Dr.
Trapnell; HL28623, to Dr. Whitsett; and Specialized Center of
Research [SCOR] grant HL56387, to both) from the National Institutes
of Health.
Source Information
From the Divisions of Pulmonary Biology (B.C.T., J.A.W.) and Neonatology (J.A.W.), Children's Hospital Medical Center, Cincinnati; and the Department of Respiratory Diseases, International Medical Center of Japan, Tokyo (K.N.).
Address reprint requests to Dr. Trapnell at the Division of Pulmonary Biology, Children's Hospital Medical Center, 3333 Burnet Ave., Cincinnati, OH 45229, or at [email protected].
References
- Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med 1958;258:1123-1142. [ISI]
- Teja K, Cooper PH, Squires JE, Schnatterly PT. Pulmonary alveolar proteinosis in four siblings. N Engl J Med 1981;305:1390-1392. [ISI][Medline]
- Nogee LM, deMello DE, Dehner LP, Colten HR. Deficiency of
pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J
Med 1993;328:406-410.
[Full Text] - Nogee LM, Dunbar AE III, Wert SE, Askin F, Hamvas A, Whitsett
JA. A mutation in the surfactant protein C gene associated with familial
interstitial lung disease. N Engl J Med 2001;344:573-579.
[Full Text] - Nogee LM, Garnier G, Dietz HC, et al. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest 1994;93:1860-1863. [ISI][Medline]
- Dirksen U, Nishinakamura R, Groneck P, et al. Human pulmonary
alveolar proteinosis associated with a defect in GM-CSF/IL-3/IL-5 receptor
common beta chain expression. J Clin Invest 1997;100:2211-2217.
[Abstract/Full Text] - deMello DE, Lin Z. Pulmonary alveolar proteinosis: a review. Pediatr Pathol Mol Med 2001;20:413-432. [CrossRef][ISI][Medline]
- Cordonnier C, Fleury-Feith J, Escudier E, Atassi K, Bernaudin JF. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. Am J Respir Crit Care Med 1994;149:788-794. [Abstract]
- Keller CA, Frost A, Cagle PT, Abraham JL. Pulmonary alveolar proteinosis in a painter with elevated pulmonary concentrations of titanium. Chest 1995;108:277-280. [Abstract]
- Ruben FL, Talamo TS. Secondary pulmonary alveolar proteinosis occurring in two patients with acquired immune deficiency syndrome. Am J Med 1986;80:1187-1190. [ISI][Medline]
- Buechner HA, Ansari A. Acute silico-proteinosis: a new pathologic variant of acute silicosis in sandblasters, characterized by histologic features resembling alveolar proteinosis. Dis Chest 1969;55:274-278. [ISI][Medline]
- Ladeb S, Fleury-Feith J, Escudier E, Tran Van Nhieu J, Bernaudin JF, Cordonnier C. Secondary alveolar proteinosis in cancer patients. Support Care Cancer 1996;4:420-426. [ISI][Medline]
- Doyle AP, Balcerzak SP, Wells CL, Crittenden JO. Pulmonary alveolar proteinosis with hematologic disorders. Arch Intern Med 1963;112:940-946. [ISI]
- Garcia Rio F, Alvarez-Sala R, Caballero P, Prados C, Pino JM, Villamor J. Six cases of pulmonary alveolar proteinosis: presentation of unusual associations. Monaldi Arch Chest Dis 1995;50:12-15. [Medline]
- Dirksen U, Hattenhorst U, Schneider P, et al. Defective
expression of granulocyte-macrophage colony-stimulating
factor/interleukin-3/interleukin-5 receptor common beta chain in children
with acute myeloid leukemia associated with respiratory failure. Blood
1998;92:1097-1103.
[Abstract/Full Text] - Ben-Dov I, Kishinevski Y, Roznman J, et al. Pulmonary alveolar proteinosis in Israel: ethnic clustering. Isr Med Assoc J 1999;1:75-78. [ISI][Medline]
- Goldstein LS, Kavuru MS, Curtis-McCarthy P, Christie HA, Farver C, Stoller JK. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest 1998;114:1357-1362. [Abstract]
- Prakash UB, Barham SS, Carpenter HA, Dines DE, Marsh HM. Pulmonary alveolar phospholipoproteinosis: experience with 34 cases and a review. Mayo Clin Proc 1987;62:499-518. [ISI][Medline]
- Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis:
progress in the first 44 years. Am J Respir Crit Care Med 2002;166:215-235.
[Abstract/Full Text] - Asamoto H, Kitaichi M, Nishimura K, Itoh H, Izumi T. Primary pulmonary alveolar proteinosis -- clinical observation of 68 patients in Japan. Nihon Kyobu Shikkan Gakkai Zasshi 1995;33:835-845. [Medline]
- Preger L. Pulmonary alveolar proteinosis. Radiology 1969;92:1291-1295. [ISI][Medline]
- Wang BM, Stern EJ, Schmidt RA, Pierson DJ. Diagnosing pulmonary
alveolar proteinosis: a review and an update. Chest 1997;111:460-466.
[Full Text] - Lee KN, Levin DL, Webb WR, Chen D, Storto ML, Golden JA. Pulmonary alveolar proteinosis: high-resolution CT, chest radiographic, and functional correlations. Chest 1997;111:989-995. [Abstract]
- Johkoh T, Itoh H, Muller NL, et al. Crazy-paving appearance at
thin-section CT: spectrum of disease and pathologic findings. Radiology
1999;211:155-160.
[Abstract/Full Text] - De Sanctis PN. Pulmonary alveolar proteinosis: a review of the findings and theories to date, with a digression on Pneumocystis carinii pneumonia. Boston Med Q 1962;13:19-35.
- Fountain FF Jr. Lactate dehydrogenase isoenzymes in alveolar proteinosis. JAMA 1969;210:1283-1283. [CrossRef][Medline]
- Fujishima T, Honda Y, Shijubo N, Takahashi H, Abe S. Increased carcinoembryonic antigen concentrations in sera and bronchoalveolar lavage fluids of patients with pulmonary alveolar proteinosis. Respiration 1995;62:317-321. [ISI][Medline]
- Minakata Y, Kida Y, Nakanishi H, Nishimoto T, Yukawa S. Change in cytokeratin 19 fragment level according to the severity of pulmonary alveolar proteinosis. Intern Med 2001;40:1024-1027. [ISI][Medline]
- Takahashi T, Munakata M, Suzuki I, Kawakami Y. Serum and
bronchoalveolar fluid KL-6 levels in patients with pulmonary alveolar
proteinosis. Am J Respir Crit Care Med 1998;158:1294-1298.
[Abstract/Full Text] - Seymour JF, Presneill JJ, Schoch OD, et al. Therapeutic
efficacy of granulocyte-macrophage colony-stimulating factor in patients
with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med
2001;163:524-531.
[Abstract/Full Text] - Kuroki Y, Takahashi H, Chiba H, Akino T. Surfactant proteins A and D: disease markers. Biochim Biophys Acta 1998;1408:334-345. [ISI][Medline]
- Selecky PA, Wasserman K, Benfield JR, Lippmann M. The clinical and physiological effect of whole-lung lavage in pulmonary alveolar proteinosis: a ten-year experience. Ann Thorac Surg 1977;24:451-461. [Abstract]
- Fraimow W, Cathcart RT, Taylor RC. Physiologic and clinical aspects of pulmonary alveolar proteinosis. Ann Intern Med 1960;52:1177-1194. [ISI]
- Pulmonary alveolar proteinosis. In: Fraser RS, M�ller NL, Colman N, Par� PD. Fraser and Par�'s diagnosis of diseases of the chest. 4th ed. Vol. 4. Philadelphia: W.B. Saunders, 1999:2700-8.
- Iyonaga K, Suga M, Yamamoto T, Ichiyasu H, Miyakawa H, Ando M. Elevated bronchoalveolar concentrations of MCP-1 in patients with pulmonary alveolar proteinosis. Eur Respir J 1999;14:383-389. [ISI][Medline]
- Schoch OD, Schanz U, Koller M, et al. BAL findings in a patient
with pulmonary alveolar proteinosis successfully treated with GM-CSF. Thorax
2002;57:277-280.
[Abstract/Full Text] - Costello JF, Moriarty DC, Branthwaite MA, Turner-Warwick M, Corrin B. Diagnosis and management of alveolar proteinosis: the role of electron microscopy. Thorax 1975;30:121-132. [Abstract]
- Kitamura T, Uchida K, Tanaka N, et al. Serological diagnosis of
idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med
2000;162:658-662.
[Abstract/Full Text] - Bedrossian CW, Luna MA, Conklin RH, Miller WC. Alveolar proteinosis as a consequence of immunosuppression: a hypothesis based on clinical and pathologic observations. Hum Pathol 1980;11:Suppl:527-535. [ISI][Medline]
- Andriole VT, Ballas M, Wilson GL. The association of nocardiosis and pulmonary alveolar proteinosis: a case study. Ann Intern Med 1963;60:266-275. [ISI]
- Supena R, Karlin D, Strate R, Cramer PG. Pulmonary alveolar proteinosis and Nocardia brain abscess: report of a case. Arch Neurol 1974;30:266-268. [CrossRef][ISI][Medline]
- Oerlemans WG, Jansen EN, Prevo RL, Eijsvogel MM. Primary cerebellar nocardiosis and alveolar proteinosis. Acta Neurol Scand 1998;97:138-141. [ISI][Medline]
- Walker DA, McMahon SM. Pulmonary alveolar proteinosis complicated by cerebral abscess: report of a case. J Am Osteopath Assoc 1986;86:447-450. [ISI][Medline]
- Pattle RE. Properties, function and origin of the alveolar lining layer. Nature 1955;175:1125-1126. [ISI]
- Weaver TE, Whitsett JA. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem J 1991;273:249-264. [ISI][Medline]
- Ikegami M, Whitsett JA, Jobe A, Ross G, Fisher J, Korfhagen T.
Surfactant metabolism in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol
Physiol 2000;279:L468-L476.
[Abstract/Full Text] - McNeely TB, Coonrod JD. Aggregation and opsonization of type A but not type B Hemophilus influenzae by surfactant protein A. Am J Respir Cell Mol Biol 1994;11:114-122. [Abstract]
- Crouch EC. Collectins and pulmonary host defense. Am J Respir
Cell Mol Biol 1998;19:177-201.
[Abstract/Full Text] - Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung
function and disease. N Engl J Med 2002;347:2141-2148.
[Full Text] - Klaus MH, Clements JA, Havel RJ. Composition of surface-active material isolated from beef lung. Proc Natl Acad Sci U S A 1961;47:1858-1859. [ISI]
- Chase WH. The surface membrane of pulmonary alveolar walls. Exp Cell Res 1959;18:15-28. [ISI][Medline]
- Pattle RE, Thomas LC. Lipoprotein composition of the film lining the lung. Nature 1961;189:844-844. [ISI]
- Larson RK, Gordinier R. Pulmonary alveolar proteinosis: report of six cases, review of the literature, and formulation of a new theory. Ann Intern Med 1965;62:292-312. [ISI]
- Ramirez J, Harlan WR Jr. Pulmonary alveolar proteinosis: nature and origin of alveolar lipid. Am J Med 1968;45:502-512. [ISI][Medline]
- Davidson JM, Macleod WM. Pulmonary alveolar proteinosis. Br J Dis Chest 1969;63:13-28. [Medline]
- Singh G, Katyal SL, Bedrossian CW, Rogers RM. Pulmonary alveolar proteinosis. Staining for surfactant apoprotein in alveolar proteinosis and in conditions simulating it. Chest 1983;83:82-86. [Abstract]
- Honda Y, Takahashi H, Shijubo N, Kuroki Y, Akino T. Surfactant protein-A concentration in bronchoalveolar lavage fluids of patients with pulmonary alveolar proteinosis. Chest 1993;103:496-499. [Abstract]
- McClenahan JB. Pulmonary alveolar proteinosis. Arch Intern Med 1974;133:284-287. [CrossRef][ISI][Medline]
- Trapnell BC, Whitsett JA. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol 2002;64:775-802. [CrossRef][ISI][Medline]
- Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994;264:713-716. [ISI][Medline]
- Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A 1994;91:5592-5596. [Abstract]
- Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem 1977;252:1998-2003. [Abstract]
- Miyatake S, Otsuka T, Yokota T, Lee F, Arai K. Structure of the chromosomal gene for granulocyte-macrophage colony stimulating factor: comparison of the mouse and human genes. EMBO J 1985;4:2561-2568. [Abstract]
- Rasko JE, Granulocyte-macrophage colony stimulating factor. In: Thomson A, ed. The cytokine handbook. 2nd ed. San Diego, Calif.: Academic Press, 1994:343-69.
- Gearing DP, King JA, Gough NM, Nicola NA. Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor. EMBO J 1989;8:3667-3676. [Abstract]
- Hayashida K, Kitamura T, Gorman DM, Arai K, Yokota T, Miyajima A. Molecular cloning of a second subunit of the receptor for human granulocyte-macrophage colony-stimulating factor (GM-CSF): reconstitution of a high-affinity GM-CSF receptor. Proc Natl Acad Sci U S A 1990;87:9655-9659. [Abstract]
- Armitage JO. Emerging applications of recombinant human
granulocyte-macrophage colony-stimulating factor. Blood 1998;92:4491-4508.
[Full Text] - Goodall GJ, Bagley CJ, Vadas MA, Lopez AF. A model for the interaction of the GM-CSF, IL-3 and IL-5 receptors with their ligands. Growth Factors 1993;8:87-97. [ISI][Medline]
- Huffman Reed JA, Rice WR, Zsengeller ZK, Wert SE, Dranoff G, Whitsett JA. GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol 1997;273:L715-L725. [ISI][Medline]
- Tarr PE. Granulocyte-macrophage colony-stimulating factor and the immune system. Med Oncol 1996;13:133-140. [ISI][Medline]
- Ruef C, Coleman DL. Granulocyte-macrophage colony-stimulating factor: pleiotropic cytokine with potential clinical usefulness. Rev Infect Dis 1990;12:41-62. [ISI][Medline]
- Nishinakamura R, Nakayama N, Hirabayashi Y, et al. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity 1995;2:211-222. [ISI][Medline]
- Robb L, Drinkwater CC, Metcalf D, et al. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A 1995;92:9565-9569. [Abstract]
- Ikegami M, Ueda T, Hull W, et al. Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. Am J Physiol 1996;270:L650-L658. [ISI][Medline]
- Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF
regulates protein and lipid catabolism by alveolar macrophages. Am J Physiol
Lung Cell Mol Physiol 2001;280:L379-L386.
[Abstract/Full Text] - Reed JA, Ikegami M, Cianciolo ER, et al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol 1999;276:L556-L563. [ISI][Medline]
- Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA.
Pulmonary epithelial cell expression of GM-CSF corrects the alveolar
proteinosis in GM-CSF-deficient mice. J Clin Invest 1996;97:649-655.
[Abstract/Full Text] - Zsengeller ZK, Reed JA, Bachurski CJ, et al. Adenovirus-mediated granulocyte-macrophage colony-stimulating factor improves lung pathology of pulmonary alveolar proteinosis in granulocyte-macrophage colony-stimulating factor-deficient mice. Hum Gene Ther 1998;9:2101-2109. [ISI][Medline]
- LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient
mice are susceptible to pulmonary group B streptococcal infection. J Clin
Invest 1999;103:563-569.
[Abstract/Full Text] - Paine R III, Preston AM, Wilcoxen S, et al. Granulocyte-macrophage
colony-stimulating factor in the innate immune response to Pneumocystis
carinii pneumonia in mice. J Immunol 2000;164:2602-2609.
[Abstract/Full Text] - Berclaz PY, Zsengeller Z, Shibata Y, et al. Endocytic
internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal
organization are coordinately regulated in alveolar macrophages by GM-CSF
and PU.1. J Immunol 2002;169:6332-6342.
[Abstract/Full Text] - Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via
PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the
IL-18/IFN-gamma-mediated molecular connection between innate and adaptive
immunity in the lung. Blood 2002;100:4193-4200.
[Abstract/Full Text] - Paine R III, Morris SB, Jin H, et al. Impaired functional
activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol
Lung Cell Mol Physiol 2001;281:L1210-L1218.
[Abstract/Full Text] - Shibata Y, Berclaz P-Y, Chroneos Z, Yoshida H, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001;15:557-567. [ISI][Medline]
- Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell 1990;61:113-124. [ISI][Medline]
- Hromas R, Orazi A, Neiman RS, et al. Hematopoietic lineage- and stage-restricted expression of the ETS oncogene family member PU.1. Blood 1993;82:2998-3004. [Abstract]
- Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 1994;265:1573-1577. [ISI][Medline]
- Celada A, Borras FE, Soler C, et al. The transcription factor PU.1 is involved in macrophage proliferation. J Exp Med 1996;184:61-69. [Abstract]
- McKercher SR, Torbett BE, Anderson KL, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J 1996;15:5647-5658. [Abstract]
- Anderson KL, Smith KA, Conners K, McKercher SR, Maki RA,
Torbett BE. Myeloid development is selectively disrupted in PU.1 null mice.
Blood 1998;91:3702-3710.
[Abstract/Full Text] - Lloberas J, Soler C, Celada A. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunol Today 1999;20:184-189. [CrossRef][ISI][Medline]
- DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage
development by graded expression of PU.1. Science 2000;288:1439-1441.
[Abstract/Full Text] - Golde DW, Territo M, Finley TN, Cline MJ. Defective lung macrophages in pulmonary alveolar proteinosis. Ann Intern Med 1976;85:304-309. [ISI][Medline]
- Harris JO. Pulmonary alveolar proteinosis: abnormal in vitro function of alveolar macrophages. Chest 1979;76:156-159. [Abstract]
- Gonzalez-Rothi RJ, Harris JO. Pulmonary alveolar proteinosis: further evaluation of abnormal alveolar macrophages. Chest 1986;90:656-661. [Abstract]
- Golde DW. Alveolar proteinosis and the overfed macrophage. Chest 1979;76:119-120. [ISI][Medline]
- Muller-Quernheim J, Schopf RE, Benes P, Schulz V, Ferlinz R. A macrophage-suppressing 40-kD protein in a case of pulmonary alveolar proteinosis. Klin Wochenschr 1987;65:893-897. [ISI][Medline]
- Nugent KM, Pesanti EL. Macrophage function in pulmonary alveolar proteinosis. Am Rev Respir Dis 1983;127:780-781. [ISI][Medline]
- Stratton JA, Sieger L, Wasserman K. The immunoinhibitory activities of the lung lavage materials and sera from patients with pulmonary alveolar proteinosis (PAP). J Clin Lab Immunol 1981;5:81-86. [ISI][Medline]
- Seymour JF, Dunn AR, Vincent JM, Presneill JJ, Pain MC.
Efficacy of granulocyte-macrophage colony-stimulating factor in acquired
alveolar proteinosis. N Engl J Med 1996;335:1924-1925.
[Full Text] - Kavuru MS, Sullivan EJ, Piccin R, Thomassen MJ, Stoller JK.
Exogenous granulocyte-macrophage colony-stimulating factor administration
for pulmonary alveolar proteinosis. Am J Respir Crit Care Med
2000;161:1143-1148.
[Abstract/Full Text] - Bewig B, Wang XD, Kirsten D, Dalhoff K, Schafer H. GM-CSF and GM-CSF beta c receptor in adult patients with pulmonary alveolar proteinosis. Eur Respir J 2000;15:350-357. [CrossRef][ISI][Medline]
- Carraway MS, Ghio AJ, Carter JD, Piantadosi CA. Detection of
granulocyte-macrophage colony-stimulating factor in patients with pulmonary
alveolar proteinosis. Am J Respir Crit Care Med 2000;161:1294-1299.
[Abstract/Full Text] - Tanaka N, Watanabe J, Kitamura T, Yamada Y, Kanegasaki S, Nakata K. Lungs of patients with idiopathic pulmonary alveolar proteinosis express a factor which neutralizes granulocyte-macrophage colony stimulating factor. FEBS Lett 1999;442:246-250. [CrossRef][ISI][Medline]
- Kitamura T, Tanaka N, Watanabe J, et al. Idiopathic pulmonary
alveolar proteinosis as an autoimmune disease with neutralizing antibody
against granulocyte/macrophage colony-stimulating factor. J Exp Med
1999;190:875-880.
[Abstract/Full Text] - Bonfield TL, Russell D, Burgess S, Malur A, Kavuru MS,
Thomassen MJ. Autoantibodies against granulocyte macrophage
colony-stimulating factor are diagnostic for pulmonary alveolar proteinosis.
Am J Respir Cell Mol Biol 2002;27:481-486.
[Abstract/Full Text] - Uchida K, Nakata K, Trapnell BC, et al. High affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood (in press).
- Hamvas A, Nogee LM, Mallory GB Jr, et al. Lung transplantation for treatment of infants with surfactant protein B deficiency. J Pediatr 1997;130:231-239. [ISI][Medline]
- Ramirez-R J, Schultz RB, Dutton RE. Pulmonary alveolar proteinosis: a new technique and rationale for treatment. Arch Intern Med 1963;112:419-431. [ISI]
- Ramirez-R J, Nyka W, McLaughlin J. Pulmonary alveolar proteinosis: diagnostic technics and observations. N Engl J Med 1963;268:165-171. [ISI]
- Wasserman K, Mason GR. Pulmonary alveolar proteinosis. In: Murray JF, Nadel JA, eds. Textbook of respiratory medicine. 2nd ed. Vol. 2. Philadelphia: W.B. Saunders, 1994:1933-46.
- Du Bois RM, McAllister WA, Branthwaite MA. Alveolar proteinosis: diagnosis and treatment over a 10-year period. Thorax 1983;38:360-363. [Abstract]
- Kariman K, Kylstra JA, Spock A. Pulmonary alveolar proteinosis: prospective clinical experience in 23 patients for 15 years. Lung 1984;162:223-231. [ISI][Medline]
- Hoffman RM, Dauber JH, Rogers RM. Improvement in alveolar macrophage migration after therapeutic whole lung lavage in pulmonary alveolar proteinosis. Am Rev Respir Dis 1989;139:1030-1032. [ISI][Medline]
- Bury T, Corhay JL, Saint-Remy P, Radermecker M. Prot�inose alv�olaire: restauration fonctionnelle macrophages alv�olaire apr�s lavage th�rapeutique. Rev Mal Respir 1989;6:373-375. [ISI][Medline]
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary
alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest
2002;122:1480-1485.
[Abstract/Full Text] - Mazzone PJ, Sullivan EJ, Piccin R, Stoller JK, Thomassen JJ, Kavuru MS. Granulocyte macrophage-colony stimulating factor therapy for pulmonary alveolar proteinosis. Am Rev Respir Dis 2000;161:A888-A888. abstract.