NEJM
Editorial
|
Thomas O. Carpenter, M.D.
Oncogenic osteomalacia has fascinated physiology-minded physicians for decades. The traditional name for this peculiar disorder connotes its classification as a paraneoplastic phenomenon. Such a characterization is a bit off the mark, however, in that the involved "neoplasm" is often (but not always) of limited clinical significance apart from its causal role in the musculoskeletal disease. Tumors responsible for oncogenic osteomalacia are usually benign rather than invasive, whereas generalized, debilitating osteomalacia and rickets are the important clinical problems for the patient. The assay for the measurement of circulating levels of fibroblast growth factor 23 (FGF-23), the development of which is described by Jonsson et al.1 in this issue of the Journal, may prove to be useful in the investigation and management of oncogenic osteomalacia.
Clinical Presentation
The clinical presentation of oncogenic osteomalacia is reminiscent of that of the more common disorder X-linked hypophosphatemia,2 which has been studied intensively and serves as the prototypic hypophosphatemic disorder. Oncogenic osteomalacia, like X-linked hypophosphatemia, is manifested by decreased mineralization of newly formed bone and the clinical findings of osteomalacia. In growing children, rachitic deformities of the growth plates occur. A useful clinical distinction between the two disorders is the patient's age at the onset of the disease: oncogenic osteomalacia is an acquired phenotype, whereas X-linked hypophosphatemia tends to become evident during the second year of life. There are exceptions to this generalization: a later onset of X-linked and autosomal dominant hypophosphatemic rickets does occur. Patients with oncogenic osteomalacia frequently present with fractures and more severe bone pain than that which occurs in X-linked hypophosphatemia and often report muscle weakness � an unusual symptom in patients with X-linked hypophosphatemia.
The characteristic hypophosphatemia results from an excessive renal loss of phosphate. The serum calcium level is usually normal, but mild hypocalcemia has been described. Elevations of serum alkaline phosphatase activity are typical, and the severity of this abnormality can exceed that seen in X-linked hypophosphatemia. Serum levels of parathyroid hormone have been variably reported as low and as elevated but are most frequently normal. Low or normal circulating levels of 1,25-dihydroxyvitamin D are observed, despite ambient hypophosphatemia, a major physiological stimulus for 1,25-dihydroxyvitamin D production; 1,25-dihydroxyvitamin D levels in oncogenic osteomalacia are often lower than those in patients with X-linked hypophosphatemia. Thus, the typical clinical and biochemical phenotype is similar to that of X-linked hypophosphatemia, but the severity of the abnormality is often greater in oncogenic osteomalacia.
Evaluation
When these clinical features occur in an older child or an adult, particularly in the absence of any relevant family history of disease, oncogenic osteomalacia should be suspected. A careful search for a tumor is an important step in the evaluation of such patients. A substantial number of deaths can be avoided by removal of the causative tumor, and in some cases, an occult cancer may be identified. Tumors are often quite small and not detectable on physical examination or routine radiography. There appears to be a propensity for these tumors to arise in the head and neck, and detailed computed tomography or magnetic resonance imaging of the sinuses and jaw areas is suggested. The occurrence of tumors at skeletal sites is not uncommon in patients with oncogenic osteomalacia. Successful localization of causative tumors with the use of indium-111 pentetreotide or octreotide scintigraphy has been demonstrated.3,4 Technetium-99 methylene diphosphonate scintigraphy has also been used but is likely to reveal uptake of isotope in areas of active osteomalacia, rather than to localize a tumor. With increasing awareness of this syndrome, and increasingly refined imaging techniques, we may discover that such tumors are not as rare as they were once thought to be.
The report by Jonsson et al. of a serum assay for the tumor-derived factor FGF-23 will most likely add to the armamentarium for the diagnosis of this condition and may help to establish whether tumors have been completely removed by surgery. Circulating levels of FGF-23, however, are increased in patients with X-linked hypophosphatemia; thus, this measure does not appear to distinguish between the two conditions. Furthermore, it is not clear whether the assay preferentially recognizes the intact FGF-23 molecule or C-terminal fragments of it. Specific identification of various molecular species may provide critical information regarding the role of FGF-23 processing in the pathogenesis of hypophosphatemia.
Treatment and Course
The clinical course of oncogenic osteomalacia is dramatically affected by removal of the tumor, which, if it is possible, is the treatment of choice. The serum phosphate level, indexes of renal phosphate wasting, and circulating levels of 1,25-dihydroxyvitamin D return to normal within hours to days after the removal of the tumor.5 Serum biochemical markers of bone turnover, such as the osteocalcin level and alkaline phosphatase activity, tend to take longer to normalize; long-term skeletal changes may require months to be corrected.6 The rapid biochemical response is remarkable, however, and the complete resolution of long-standing symptoms is a major relief to patients. Rarely in the treatment of metabolic diseases does a physician encounter such a complete reversal of debilitating symptoms � a truly gratifying experience.
If a tumor is not found, or if an identified tumor is not resectable, it is recommended that vitamin D metabolites (preferably calcitriol) and oral phosphate salts be used in a manner similar to that used in the treatment of X-linked hypophosphatemia.2 This treatment usually provides some benefit, although it does not result in a complete clinical response. Long-term treatment with intravenous phosphate, together with oral calcium and vitamin D, has been successfully used when oral phosphate salts were not tolerated.7 This therapy can result in bloodstream infection related to a central venous catheter and thus should be used only when the promise of a benefit is greater than the risk of catheter-related complications. As a short-term treatment, octreotide therapy rapidly corrected the serum phosphate level and alkaline phosphatase activity in a patient with a tumor that had been detected by octreotide scintigraphy.4
Most tumors associated with oncogenic osteomalacia are benign, consisting of mesenchymal cells or mixed connective tissue. Soft-tissue tumors are often vascular, with abundant spindle or giant cells, and are frequently reported as hemangiopericytomas. Tumors that arise in bone are usually classified as chondroblastic�osteoblastic, ossifying, or nonossifying fibromas, as categorized by Weidner and Santa Cruz.8 Occasional malignant tumors have been reported.9 According to one report, osteosarcoma was discovered in a patient who had had oncogenic osteomalacia for many years before its source was identified.10 Other patients have had recurrent disease that has progressed and become fatal. Thus, it is highly recommended that the surgeon establish a margin of resection that is free of tumor, particularly in the case of tumors with substantial atypia or mitotic elements.
Pathophysiology and Candidate Mediators
Despite improvements in our understanding of oncogenic osteomalacia and X-linked hypophospha-temia, the puzzle has not been completely solved. Loss of function of the PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) protease determines the X-linked hypophosphatemia phenotype, but how this occurs remains a mystery.2 Specific mutations of FGF-23 result in autosomal dominant hypophosphatemic rickets11; this may be due to a mutation-induced inhibition of processing.12 The injection of FGF-23�transfected cells into mice results in the characteristic features of oncogenic osteomalacia.13 On the other hand, other factors identified from tumors responsible for oncogenic osteomalacia have been implicated in phosphate homeostasis; these factors include frizzled-related protein 4, matrix extracellular phosphoglycoprotein, and unidentified substances with small molecular weights.14 Figure 1 shows the central role of FGF-23 in the proposed mechanisms underlying hypophosphatemic disorders.
|
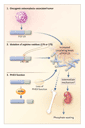
A number of important questions remain. Does PHEX degrade FGF-23, thereby accounting for increased levels in patients with X-linked hypophosphatemia? Is FGF-23 the direct mediator responsible for impaired transport of phosphate by the renal tubules and the impaired regulation of vitamin D metabolism, or does another factor in a complex cascade of signals perform this function? Is the bone disease simply a result of prolonged hypophosphatemia, or does FGF-23 or another tumor-derived substance have a direct effect on the skeleton? What are the roles of frizzled-related protein 4 and matrix extracellular phosphoglycoprotein? The finding that increased circulating FGF-23 levels occur in X-linked hypophosphatemia as well as in oncogenic osteomalacia argues that these two disorders do share a common pathophysiological pathway. Further clinical investigation made possible by this new assay may shed further light on the complex pathophysiology involved.
Dr. Carpenter reports having received consulting fees from
Merck and Genzyme and a grant from CuraGen.
Source Information
From the Section of Pediatric Endocrinology, Yale University School of Medicine, New Haven, Conn.
References
- Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth
factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J
Med 2003;348:1656-1663.
[Abstract/Full Text] - Carpenter TO. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin North Am 1997;44:443-466. [ISI][Medline]
- Nguyen BD, Wang EA. Indium-111 pentetreotide scintigraphy of mesenchymal tumor with oncogenic osteomalacia. Clin Nucl Med 1999;24:130-131. [CrossRef][ISI][Medline]
- Seufert J, Ebert K, M�ller J, et al. Octreotide therapy for
tumor-induced osteomalacia. N Engl J Med 2001;345:1883-1888.
[Full Text] - Agus ZS. Oncogenic hypophosphatemic osteomalacia. Kidney Int 1983;24:113-123. [ISI][Medline]
- Shane E, Parisien M, Henderson JE, et al. Tumor-induced osteomalacia: clinical and basic studies. J Bone Miner Res 1997;12:1502-1511. [ISI][Medline]
- Yeung SJ, McCutcheon IE, Schultz P, Gagel RF. Use of long-term
intravenous phosphate infusion in the palliative treatment of tumor-induced
osteomalacia. J Clin Endocrinol Metab 2000;85:549-555.
[Abstract/Full Text] - Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors: a polymorphous group causing osteomalacia or rickets. Cancer 1987;59:1442-1454. [ISI][Medline]
- Drezner MK. Tumor-induced osteomalacia. In: Favus MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 1999:331-7.
- Cheng CL, Ma J, Wu PC, Mason RS, Posen S. Osteomalacia secondary to osteosarcoma: a case report. J Bone Joint Surg Am 1989;71:288-292. [Medline]
- The ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000;26:345-348. [CrossRef][ISI][Medline]
- White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 2001;60:2079-2086. [CrossRef][ISI][Medline]
- Shimada T, Mizutani S, Muto T, et al. Cloning and
characterization of FGF23 as a causative factor of tumor-induced
osteomalacia. Proc Natl Acad Sci U S A 2001;98:6500-6505.
[Abstract/Full Text] - Schiavi SC, Moe OW. Phosphatonins: a new class of phosphate-regulating proteins. Curr Opin Nephrol Hypertens 2002;11:423-430. [CrossRef][ISI][Medline]